























علم الكيمياء

تاريخ الكيمياء والعلماء المشاهير
التحاضير والتجارب الكيميائية
المخاطر والوقاية في الكيمياء
اخرى
مقالات متنوعة في علم الكيمياء
كيمياء عامة
الكيمياء التحليلية
مواضيع عامة في الكيمياء التحليلية
التحليل النوعي والكمي
التحليل الآلي (الطيفي)
طرق الفصل والتنقية
الكيمياء الحياتية
مواضيع عامة في الكيمياء الحياتية
الكاربوهيدرات
الاحماض الامينية والبروتينات
الانزيمات
الدهون
الاحماض النووية
الفيتامينات والمرافقات الانزيمية
الهرمونات
الكيمياء العضوية
مواضيع عامة في الكيمياء العضوية
الهايدروكاربونات
المركبات الوسطية وميكانيكيات التفاعلات العضوية
التشخيص العضوي
تجارب وتفاعلات في الكيمياء العضوية
الكيمياء الفيزيائية
مواضيع عامة في الكيمياء الفيزيائية
الكيمياء الحرارية
حركية التفاعلات الكيميائية
الكيمياء الكهربائية
الكيمياء اللاعضوية
مواضيع عامة في الكيمياء اللاعضوية
الجدول الدوري وخواص العناصر
نظريات التآصر الكيميائي
كيمياء العناصر الانتقالية ومركباتها المعقدة
مواضيع اخرى في الكيمياء
كيمياء النانو
الكيمياء السريرية
الكيمياء الطبية والدوائية
كيمياء الاغذية والنواتج الطبيعية
الكيمياء الجنائية
الكيمياء الصناعية
البترو كيمياويات
الكيمياء الخضراء
كيمياء البيئة
كيمياء البوليمرات
مواضيع عامة في الكيمياء الصناعية
الكيمياء التناسقية
الكيمياء الاشعاعية والنووية

Six-Membered Rings
 المؤلف:
المؤلف:
 المصدر:
المصدر:
 الجزء والصفحة:
الجزء والصفحة:
 21-12-2015
21-12-2015
 8153
8153




+
-
20
Six-Membered Rings
Properties
The chemical reactivity of the saturated members of this class of heterocycles: tetrahydropyran, thiane and piperidine, resemble that of acyclic ethers, sulfides, and 2º-amines, and will not be described here. 1,3-Dioxanes and dithianes are cyclic acetals and thioacetals. These units are commonly used as protective groups for aldehydes and ketones, as well as synthetic intermediates, and may be hydrolyzed by the action of aqueous acid. The reactivity of partially unsaturated compounds depends on the relationship of the double bond and the heteroatom (e.g. 3,4-dihydro-2H-pyran is an enol ether).
Fully unsaturated six-membered nitrogen heterocycles, such as pyridine, pyrazine, pyrimidine and pyridazine, have stable aromatic rings. Oxygen and sulfur analogs are necessarily positively charged, as in the case of 2,4,6-triphenylpyrylium tetrafluoroborate.
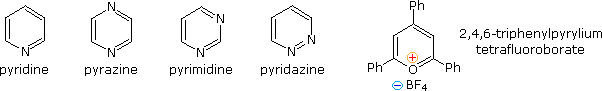
From heat of combustion measurements, the aromatic stabilization energy of pyridine is 21 kcal/mole. The resonance description drawn at the top of the following diagram includes charge separated structures not normally considered for benzene. The greater electronegativity of nitrogen (relative to carbon) suggests that such canonical forms may contribute to a significant degree. Indeed, the larger dipole moment of pyridine compared with piperidine supports this view. Pyridine and its derivatives are weak bases, reflecting the sp2 hybridization of the nitrogen. From the polar canonical forms shown here, it should be apparent that electron donating substituents will increase the basicity of a pyridine, and that substituents on the 2 and 4-positions will influence this basicity more than an equivalent 3-substituent. The pKa values given in the table illustrate a few of these substituent effects. Methyl substituted derivatives have the common names picoline (methyl pyridines), lutidine (dimethyl pyridines) and collidine (trimethyl pyridines). The influence of 2-substituents is complex, consisting of steric hindrance and electrostatic components. 4-Dimethylaminopyridine is a useful catalyst for acylation reactions carried out in pyridine as a solvent. At first glance, the sp3 hybridized nitrogen might appear to be the stronger base, but it should be remembered that N,N-dimethylaniline has a pKa slightly lower than that of pyridine itself. Consequently, the sp2 ring nitrogen is the site at which protonation occurs.
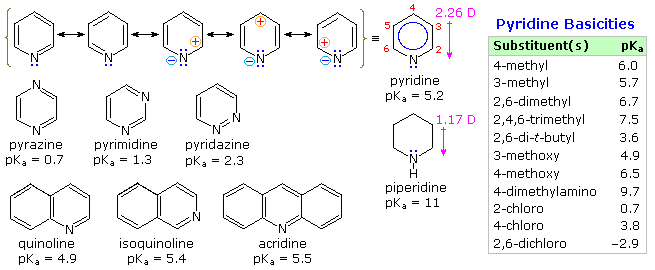
The diazines pyrazine, pyrimidine and pyridazine are all weaker bases than pyridine due to the inductive effect of the second nitrogen. However, the order of base strength is unexpected. A consideration of the polar contributors helps to explain the difference between pyrazine and pyrimidine, but the basicity of pyridazine seems anomalous. It has been suggested that electron pair repulsion involving the vicinal nitrogens destabilizes the neutral base relative to its conjugate acid.
Electrophilic Substitution of Pyridine
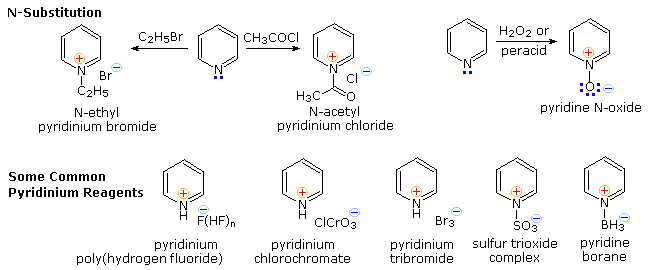
Pyridine is a modest base (pKa=5.2). Since the basic unshared electron pair is not part of the aromatic sextet, as in pyrrole, pyridinium species produced by N-substitution retain the aromaticity of pyridine. As shown below, N-alkylation and N-acylation products may be prepared as stable crystalline solids in the absence of water or other reactive nucleophiles. The N-acyl salts may serve as acyl transfer agents for the preparation of esters and amides. Because of the stability of the pyridinium cation, it has been used as a moderating component in complexes with a number of reactive inorganic compounds. Several examples of these stable and easily handled reagents are shown at the bottom of the diagram. The poly(hydrogen fluoride) salt is a convenient source of HF for addition to alkenes and conversion of alcohols to alkyl fluorides, pyridinium chlorochromate (PCC) and its related dichromate analog are versatile oxidation agents and the tribromide salt is a convenient source of bromine. Similarly, the reactive compounds sulfur trioxide and diborane are conveniently and safely handled as pyridine complexes.
Amine oxide derivatives of 3º-amines and pyridine are readily prepared by oxidation with peracids or peroxides, as shown by the upper right equation. Reduction back to the amine can usually be achieved by treatment with zinc (or other reactive metals) in dilute acid.
From the previous resonance description of pyridine, we expect this aromatic amine to undergo electrophilic substitution reactions far less easily than does benzene. Furthermore, as depicted above by clicking on the diagram, the electrophilic reagents and catalysts employed in these reactions coordinate with the nitrogen electron pair, exacerbating the positive charge at positions 2,4 & 6 of the pyridine ring. Three examples of the extreme conditions required for electrophilic substitution are shown on the left. Substituents that block electrophile coordination with nitrogen or reduce the basicity of the nitrogen facilitate substitution, as demonstrated by the examples in the blue-shaded box at the lower right, but substitution at C-3 remains dominant. Activating substituents at other locations also influence the ease and regioselectivity of substitution. By clicking on the diagram a second time, three examples will shown on the left. The amine substituent in the upper case directs the substitution to C-2, but the weaker electron donating methyl substituent in the middle example cannot overcome the tendency for 3-substitution. Hydroxyl substituents at C-2 and C-4 tautomerize to pyridones, as shown for the 2-isomer at the bottom left.
Pyridine N-oxide undergoes some electrophilic substitutions at C-4 and others at C-3. The coordinate covalent N–O bond may exert a push-pull influence, as illustrated by the two examples on the right. Although the positively charged nitrogen alone would have a strong deactivating influence, the negatively charged oxygen can introduce electron density at C-2, C-4 & C-6 by π-bonding to the ring nitrogen. This is a controlling factor in the relatively facile nitration at C-4. However, if the oxygen is bonded to an electrophile such as SO3, the resulting pyridinium ion will react sluggishly and preferentially at C-3.
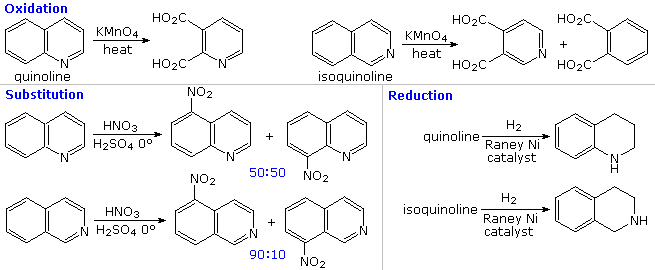
The fused ring heterocycles quinoline and isoquinoline provide additional evidence for the stability of the pyridine ring. Vigorous permanganate oxidation of quinoline results in predominant attack on the benzene ring; isoquinoline yields products from cleavage of both rings. Note that naphthalene is oxidized to phthalic acid in a similar manner. By contrast, the heterocyclic ring in both compounds undergoes preferential catalytic hydrogenation to yield tetrahydroproducts. Electrophilic nitration, halogenation and sulfonation generally take place at C-5 and C-8 of the benzene ring, in agreement with the preceding description of similar pyridine reactions and the kinetically favored substitution of naphthalene at C-1 (α) rather than C-2 (β).
Other Reactions of Pyridine
Thanks to the nitrogen in the ring, pyridine compounds undergo nucleophilic substitution reactions more easily than equivalent benzene derivatives. In the following diagram, reaction 1 illustrates displacement of a 2-chloro substituent by ethoxide anion. The addition-elimination mechanism shown for this reaction is helped by nitrogen's ability to support a negative charge. A similar intermediate may be written for substitution of a 4-halopyridine, but substitution at the 3-position is prohibited by the the failure to create an intermediate of this kind. The two Chichibabin aminations in reactions 2 and 3 are remarkable in that the leaving anion is hydride (or an equivalent). Hydrogen is often evolved in the course of these reactions. In accord with this mechanism, quinoline is aminated at both C-2 and C-4.
Addition of strong nucleophiles to N-oxide derivatives of pyridine proceed more rapidly than to pyridine itself, as demonstrated by reactions 4 and 5. The dihydro-pyridine intermediate easily loses water or its equivalent by elimination of the –OM substituent on nitrogen.
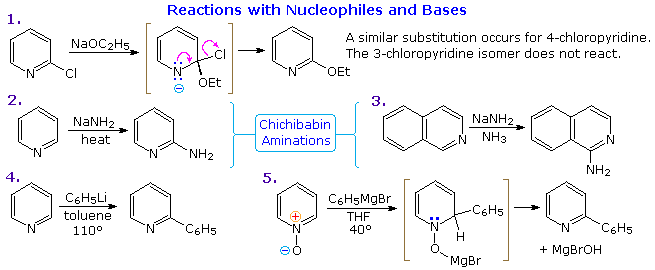
By clicking on the above diagram, five additional examples of base or nucleophile reactions with substituted pyridine will be displayed. Because the pyridine ring (and to a greater degree the N-oxide ring) can support a negative charge, alkyl substituents in the 2- and 4-locations are activated in the same fashion as by a carbonyl group. Reactions 6 and 7 show alkylation and condensation reactions resulting from this activation. Reaction 8 is an example of N-alkylpyridone formation by hydroxide addition to an N-alkyl pyridinium cation, followed by mild oxidation. Birch reduction converts pyridines to dihydropyridines that are bis-enamines and may be hydrolyzed to 1,5-dicarbonyl compounds. Pyridinium salts undergo a one electron transfer to generate remarkably stable free radicals. The example shown in reaction 9 is a stable (in the absence of oxygen), distillable green liquid. Although 3-halopyridines do not undergo addition-elimination substitution reactions as do their 2- and 4-isomers, the strong base sodium amide effects amination by way of a pyridyne intermediate. This is illustrated by reaction 10. It is interesting that 3-pyridyne is formed in preference to 2-pyridyne. The latter is formed if C-4 is occupied by an alkyl substituent. The pyridyne intermediate is similar to benzyne.
Reference
http://www2.chemistry.msu.edu/faculty/reusch/VirtTxtJml/heterocy.htm#top1
 الاكثر قراءة في مواضيع عامة في الكيمياء العضوية
الاكثر قراءة في مواضيع عامة في الكيمياء العضوية
 اخر الاخبار
اخر الاخبار
اخبار العتبة العباسية المقدسة

الآخبار الصحية


 قسم الشؤون الفكرية يصدر كتاباً يوثق تاريخ السدانة في العتبة العباسية المقدسة
قسم الشؤون الفكرية يصدر كتاباً يوثق تاريخ السدانة في العتبة العباسية المقدسة "المهمة".. إصدار قصصي يوثّق القصص الفائزة في مسابقة فتوى الدفاع المقدسة للقصة القصيرة
"المهمة".. إصدار قصصي يوثّق القصص الفائزة في مسابقة فتوى الدفاع المقدسة للقصة القصيرة (نوافذ).. إصدار أدبي يوثق القصص الفائزة في مسابقة الإمام العسكري (عليه السلام)
(نوافذ).. إصدار أدبي يوثق القصص الفائزة في مسابقة الإمام العسكري (عليه السلام)



















