
آخر المواضيع المضافة

 النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية
النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية| Adenylylation |


|
|
Read More
Date: 27-12-2015
Date: 18-5-2016
Date: 28-3-2021
|
Adenylylation
Adenylylation is the process in which adenosine-5′-monophosphate (AMP) is covalently attached to a protein, nucleic acid, or small molecule via a phosphodiester or phosphoramidate linkage. Most often, the AMP is derived from ATP, but in some bacterial adenylylation reactions NADP+ is the source. Similarly, deadenylylation is the process in which AMP is removed from the adenylylated molecule. The adenylylation/deadenylylation processes may provide regulatory control of enzyme activity, contribute to intermediate steps in individual enzymatic reaction mechanisms, or occur as intermediate steps along the biosynthetic pathway of cofactors. In this sense, adenylylation is analogous to phosphorylation, sulfation, methylation, and other intracellular covalent modification reactions for which multiple functions exist. Adenylylation occurs in a wide range of organisms, including bacteria, yeast, and mammals, although it is less common than many other post-translational modification reactions as a source of enzyme regulation. The general chemical reaction for ATP-dependent adenylylation is shown in Figure 1.

Figure 1. General reaction scheme for ATP-dependent adenylylation. R = O or N. The a, b, and g phosphorous atoms are labeled.
It is important to distinguish adenylylation from the related processes of phosphorylation, adenylation, and ADP-ribosylation:
1. Phosphorylation is readily distinguished from each of the other processes by the lack of incorporation of sugar or adenine in the acceptor.
2. Adenylation results in covalent attachment of ADP via the b-phosphoryl group. Relatively few examples of adenylation are known, and they appear to be limited to adenylation of carbohydrates to yield, for example, glucose-1-ADP.
3. ADP-ribosylation results in a covalent bond between the ribose moiety of NADPH and an acceptor.
These covalent modification reactions also contribute to multiple biological functions, including regulation of enzymatic activity. Analytically, adenylation may be distinguished from adenylylation with the appropriate radiolabeled substrates. Specifically, a-32P-ATP, but not b-32P-ATP or g-32P-ATP, will yield radiolabeled acceptor if adenylylation occurs. Typically, ATP that is radiolabeled with 3H or 13C in the adenine moiety is also used, in separate experiments, to confirm that label incorporated into the acceptor is not a result of phosphorylation, without adenylylation. An additional criterion that is often applied to distinguish adenylylation from phosphorylation is its sensitivity to phosphodiesterases. Phosphodiesterases will cleave the AMP from an adenylylated substrate, but they will not hydrolyze phosphate from phosphorylated protein. Specific examples of molecules that are adenylylated are discussed separately below.
1. Glutamine Synthetase
In all organisms, glutamine synthetase (GS) plays a critical role in intermediary metabolism by catalyzing the ATP-dependent condensation of ammonia with glutamate, to yield glutamine. It was discovered by Stadtman and co-workers (1, 2) that Escherichia coli glutamine synthetase exhibited dramatically different kinetic properties when isolated from cultures grown in nitrogen-rich vs. nitrogen-starved media. They demonstrated that the differences in enzymatic activity of GS corresponded to the presence or absence of covalently attached adenylyl groups. Since their initial findings, bacterial GS and associated regulatory enzymes have provided a paradigm for understanding biological regulation of nitrogen assimilation in some prokaryotic organisms. Comparison of the molecular details of adenylylation-dependent regulation of prokaryotic GS remains an active research area. Mammalian and plant GS, in contrast, are not regulated by adenylylation.
Regulation of glutamine biosynthesis is characterized most thoroughly for E. coli and includes a complex bicyclic cascade that controls the adenylylation of Tyr397 on the surface of GS. Bacterial GS are dodecameric oligomers with two face-to-face hexameric rings. In the subset of bacterial strains that regulate GS activity via adenylylation, the adenylylation state of GS may vary from 0 to 12 AMPs/GS dodecamer, and the enzymatic activity decreases with increasing extent of adenylylation. The regulatory cascade is summarized in Figure 2. It includes the adenylyl transferase (ATase), a signal transduction enzyme (PII), and a uridylyl transferase (UTase) enzyme that responds directly to nitrogen levels. When the nitrogen levels are low, UTase uridylylates PII at Tyr51 to form PII-UMP. PII-UMP stimulates the deadenylylation activity of ATase, to decrease the proportion of GS in the adenylylated form and thus to increase the rate at which nitrogen is “fixed” in the amino acid glutamine, derived from glutamate by GS. When nitrogen levels are high, the UTase cleaves the UMP from PII-UMP to generate PII. Unmodified PII stimulates the adenylylation activity of ATase, to increase the adenylylation state of GS and reduce its efficiency in converting glutamate to glutamine. The complexity of these regulatory cascades underscores the importance of adenylylation as a mechanism for controlling nitrogen metabolism in some prokaryotes.

Figure 2. Bicyclic cascade for regulation of E. coli glutamine synthetase (GS). ATase catalyzes the adenylylation/deadenylylation of GS; PII stimulates the adenylylation activity, and uridylylated PII (PII-UMP) stimulates the deadenylylation activity. Uridylylation of PII is catalyzed by UTase. UTase uridylylates PII when nitrogen levels are low, and it hydrolyzes the uridylyl group from PII-UMP when nitrogen levels are high.
The structural basis for the decrease in enzymatic activity of E. coli GS upon adenylylation is not known. Although X-ray crystallography structures are available for the unadenylylated GS from Salmonella typhimurium (3, 4), no structure for an adenylylated GS has been determined. Spectroscopic and kinetic analyses indicate that adenylylation causes an increase in the Km (Michaelis constant) for substrates and for the metal cofactors (Mg2+ or Mn2+) that are required at each GS active site, although no specific interactions between the adenylyl group and protein residues have been identified (5-7). On the basis of many biophysical criteria and hydrodynamic volume properties, the adenylylated and unadenylylated forms of GS do not differ greatly in conformation (8, 9). Presumably, adenylyl groups attached at Tyr397 of each subunit in dodecameric GS induce subtle changes in the local conformation of the active sites to which they are adjacent. Spectroscopic methods have suggested that the adenine moiety of AMP attached to each subunit within the hexameric ring structure is highly dynamic, and it collides by diffusion with the subunit adjacent to it (10, 11). Thus, specific hydrogen bonds or electrostatic interactions between the adenylyl groups and protein residues may be limited to the phosphate and ribose moieties of AMP. Alternatively, AMP may simply block the enzyme active sites sterically.
Based on sequence comparisons of GS from numerous prokaryotes, and on mutational analysis of the E. coli and Anabaena 7120 GS, a minimal consensus sequence including the Tyr 397 to be adenylylated and a properly positioned proline residue are required for efficient adenylylation by ATase. These two residues are indicated in bold in the local sequence of E. coli GS: Met-Asp-Lys-Asn-Leu-Tyr-Asp-Leu-Pro-Pro-Glu-Glu-Ser-Lys. Individual mutation of several residues in this sequence has negligible or modest effects on the rate of adenylylation by ATase. In contrast, replacement of the bold proline nearly abolishes the ATase-catalyzed adenylylation, and substitution of serine by proline at the analogous position in the Anabaena 7120 is sufficient to make this GS a substrate for ATase from E. coli (12, 13).
The E. coli Atase has been characterized genetically and biochemically; it is a constitutively expressed 945 amino acid residue protein (~115 kDa) that contains two highly homologous domains (8, 14). Genetically engineered N- and 3C-terminal domains that have been expressed separately and purified are both catalytically active. However, the two nonoverlapping constructs exhibit different catalytic activities. The N-terminal domain (residues 1 to 423) catalyzes deadenylylation of GS,
requires PII-UMP, and is inhibited by PII. In contrast, the C-terminal domain (residues 425 to 945) catalyzes the adenylylation of GS and is not regulated by PII or PII-UMP. It is striking that PII is required for the adenylylation activity of the full-length ATase, whereas the C-terminal domain by itself is unresponsive to PII or PII-UMP. Presumably, PII and PII-UMP regulate the adenylylation reaction of the C-terminal domain by interacting with the N-terminal domain, or the full-length construct is required to maintain the structural integrity of this regulatory site. In addition, glutamine activates the adenylylation reaction of the C-terminal domain. It also is noteworthy that the adenylylation and deadenylylation reactions catalyzed by ATase are not the reverse of each other. Deadenylylation requires inorganic phosphate (Pi) rather than PPi, and the reaction product is ADP
rather than ATP. In effect, this deadenylylation reaction may be considered formally as transfer of an adenylyl group from GS to Pi, or the adenylylation of phosphate anion. Although the two functional domains of ATase exhibit significant homology at the amino acid level, they have clearly distinct functional properties. The structural basis for these differences remains unknown.
2. DNA Ligases, RNA Ligases, and Related Enzymes
DNA ligases and RNA ligases catalyze the formation of phosphodiester bonds at single strand breaks with adjacent 3′-hydroxyl and 5′-phosphate termini in DNA or RNA, respectively. For bacterial, mammalian, and virus ligases, the first step in the catalytic cycle is the adenylylation of an active-site e-amino group of a lysine side chain (Fig. 3). Mammalian and virally encoded ligases utilize ATP to adenylylate the lysine, whereas bacterial ligases exploit NADP+ . A consensus sequence found in ligases from each of these sources that includes the adenylylated Lys, in bold, is -Lys-X-(Asp/Asn)-Gly- (15, 16). The phosporamidate bond formed upon adenylylation of the lysine residue is more stable toward hydrolysis than is the phosphoester bond formed upon adenylylation of tyrosine residues or hydroxyl groups of aminoglycosides. Interestingly, a similar consensus sequence is found in messenger RNA-capping enzymes that transfer guanylylate to the 5′-terminus of mRNA, followed by methylation to generate the mature RNA message.
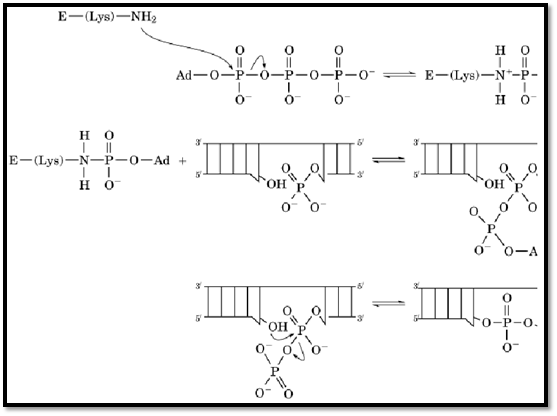
Figure 3. Mechanism for DNA and RNA ligases. The catalytic reaction begins with ATP-dependent adenylylation of the adenine; E, a ligase.
Functionally related RNA 3′-phospho-cyclases have been identified, and they appear to be present in mammals, yeast, enteric bacteria, and archaebacteria. This wide distribution suggests that these enzymes have a critical role, but no physiological function has been determined. As part of the catalytic cycle, the 3′-phosphoryl group on the terminal nucleotide of transfer RNA or small nuclear RNA (snRNA) undergoes ATP-dependent adenylylation, to generate a phosphodiester bond. This intermediate is attacked subsequently by the 2′-hydroxyl group to release AMP and produce a terminal cyclic phosphate (17). As with the ligase-catalyzed reactions, this cyclization proceeds via an initial adenylylation reaction.
3. Other Mammalian Proteins
On the basis of radiolabeling experiments, it has been suggested that plasma membrane proteins from liver or parotid glands of rats are adenylylated (18, 19). No function has been ascribed to any of these proteins. The hepatic proteins are glycosylated, and their adenylylation is inhibited by lectins (20). An interesting possibility is that the adenylyl group is attached to these proteins via their carbohydrates. The same proteins are phosphorylated, and it has been suggested that some protein kinases may be capable of catalyzing adenylylation, in addition to phosphorylation. Further studies that demonstrate covalent attachment of adenylyl groups unambiguously are required, and the physiological function for adenylylation of these proteins remains unclear.
4. Aminoglycoside Antibiotics
Several aminoglycoside antibiotics, including tobramycin, gentamycin, and kanamycin, are adenylylated by bacterial nucleotidyl transferases, and this contributes to antibiotic resistance in some strains. The enzymes responsible are not specific for ATP, and they also utilize GTP or UTP, thus resulting in guanylylation or uridylylation as well. These nucleotidyl transferases are plasmid-encoded. The adenylylation (nucleotidylylation) site on various aminoglycoside antibiotics differs, but it is most frequently either (a) the 2′- or the 3′-hydroxyl of the 3-aminoglucose ring or (b) the 4′-hydroxyl of the 6-aminoglucose ring. Based on the X-ray crystallography structure of kanamycin nucleotidyl transferase (21), the catalytic mechanism has been proposed to be an in-line displacement of PPi by the hydroxyl group of the aminoglycoside, with an active-site glutamate residue acting as a general base. The presence of only a few contacts between the adenine ring of ATP and the nucleotidyl transferase active site is consistent with the lack of specificity for the nucleotide substrate.
5. Adenosine-5-Phosphosulfate
An additional example of adenylylation is provided by the biosynthetic pathway of the cofactor 3′-phospho-adenosine-5′-phosphosulfate (PAPS), which is summarized in Figure 4. This cofactor is used by several sulfotransferases in the sulfation of catechols, phenols, and other alcohols. An intermediate formed en route to PAPS is adenosine-5′-phosphosulfate (APS). APS is formed by ATP sulfurylase, in an adenylylation reaction that requires ATP and sulfate ion, to yield APS and PPi. The formation of APS is partially rate-limiting in PAPS biosynthesis, and deficiency of ATP-sulfurylase may cause impaired sulfation of proteoglycans required for maintenance of extracellular matrix, cartilage, and connective tissues. Thus, the adenylylation reaction catalyzed by this enzyme may have direct clinical impact, manifested as chondrodysplasias, if it operates inadequately (22).

Figure 4. Biosynthesis of 3′-phospho-adenosine-5′-phosphosulfate (PAPS). The first step in the biosynthetic pathway is adenylylation of sulfate to generate adenosine-5′-phosphosulfate (APS).
References
1. B. M. Shapiro, H. S. Kingdon, and E. R. Stadtman (1967) Proc. Natl. Acad. Sci. USA 58, 642649- .
2. B. M. Shapiro and E. R. Stadtman (1968) J. Biol. Chem. 243, 3769–3771.
3. R. J. Almassy, C. A. Janson, R. Hamlin, N. H. Xuong, and D. Eisenberg (1986) Nature 323. 304–309,
4. M. M. Yamashita, R. J. Almassy, C. A. Janson, D. Lasek, and D. Eisenberg (1989) J. Biol. Chem. 264, 17681–17689.
5. A. Ginsburg, J. Yeh, S. B. Hennig, and M. D. Denton (1970) Biochemistry 9, 633–648.
6. L. M. Abell and J. J. Villafranca (1991) Biochemistry 30, 1413–1418.
7. L. P. Reynaldo, J. J. Villafranca, and W. DeW. Horrocks Jr. (1996) Prot. Sci. 5, 2532–2544.
8. S. B. Hennig and A. Ginsburg (1971) Arch. Biochem. Biophys. 144, 611–627.
9. W. M. Atkins (1994) Biochemistry 33, 14965–14973.
10. J. J. Villafranca, S. G. Rhee, and P. B. Chock (1978) Proc. Natl. Acad. Sci. USA 75, 12551259– .
11.W. M. Atkins, B. M. Cader, J. Hemmingsen, and J. J. Villafranca (1993) Protein Sci. 2, 800813- .
12. E. D. Longton, B. M. Cader, J. Hemmingsen, and J. J. Villafranca (1992) In Biosynthesis and Molecular Regulation of Amino Acids in Plants (B. K. Singh, H. E. Flores, and J. C. Shannon, eds.), American Society of Plant Physiologists, Rockville, MD, pp. 59–68.
13. J. Hemmingsen (1991) Doctoral dissertaion, The Pennsylvania State University, University Park, PA.
14. R. Jaggi, W. C. van Heeswijk, H. V. Westerhoff, D. Ollis, and S. G. Vasudevan (1997) EMBO J. 16, 5562–5571.
15. T. Lindahl and D. E. Barnes (1992) Annu. Rev. Biochem. 61, 251–281.
16. A. E. Tomkinson, N. F. Totty, M. Ginsburg, and T. Lindahl (1991) Proc. Natl. Acad. Sci. USA88, 400–404 .
17. P. Genschik, E. Billy, M. Swianiewicz, and W. Filipowicz (1997) EMBO J. 16, 2955–2967.
18. E. San Jose, A. Benguria, and A. Villalobo (1990) J. Biol. Chem. 265, 20653–20661.
19. M. Hara-Yokoyama, H. Sugiya, and S. Furuyama. (1994) Int. J. Biochem. 26, 1103–1109.
20. E. San Jose, H. J. E. Villalobo, H-J. Gabius, and A. Villalobo (1993) Biol. Chem. Hoppe-Seyler374, 133–141374 .
21. L. C. Pedersen, M. M. Benning, and H. M. Holden (1995) Biochemistry 34, 13305–13311.
22. A. Superti-Furga (1994) Am. J. Hum. Genet. 55, 1137–1145.

|
|
|
|
تفوقت في الاختبار على الجميع.. فاكهة "خارقة" في عالم التغذية
|
|
|
|
|
|
|
أمين عام أوبك: النفط الخام والغاز الطبيعي "هبة من الله"
|
|
|
|
|
|
|
قسم شؤون المعارف ينظم دورة عن آليات عمل الفهارس الفنية للموسوعات والكتب لملاكاته
|
|
|
















