آخر المواضيع المضافة

 النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية
النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية| The Human Genome Project |


|
|
Read More
Date: 11-11-2015
Date: 10-11-2015
Date: 10-11-2015
|
The Human Genome Project
In February 2001, two research groups, a biotechnology company and the other a publicly funded consortium published the draft sequence of the human genome. Accounting for just over 90% of the euchromatic portion of the 3 megabase haploid genome, this represents the fruits of a decade of intensive efforts to map and sequence the entire human genome, in the process dubbed the “Human Genome Project”.
HISTORY
The idea of sequencing the entire human genome was first proposed in the mid-eighties at scientific meetings sponsored by the US Department of Energy. The US National Research Council in 1988 recommended a broader program including the following aims: (a) The generation of detailed genetic and physical maps of the human genome, (b) sequencing of the genomes of model organisms (bacteria, yeast, worms, flies and mouse), (c) the development of technologies to support these activities, and (d) research into the ethical, legal and social issues (ELSIs) raised by human genome research. The human genome project was launched in the US in 1990 as a joint effort of the DOE and the National Institutes of Health; the plan was to finish sequencing the human genome by 2005 with an estimated budget of 3 billion dollars. By 1991 the International Human Genome Project was underway with collaborations between various organisations in the US, UK, France, Japan, European Community, and later by Germany and Japan. The Human Genome Organisation (HUGO) was founded to coordinate this international effort. By 1995, significant progress had been made in generation of the genetic and physical maps of the human genome, and in the large-scale sequencing of the yeast and worm genomes, as well as targeted regions of the human genome. In 1998, Celera Genomics [formed by a merger of Applied Biosystems and The Institute of Genomic Research (TIGR)] announced that it would independently sequence the entire human genome over a 3-year period. In February 2001, the International Human Genome Sequencing Consortium and Celera Genomics independently published the draft version of the complete human genome sequence in the journals Nature (1) and Science (2), respectively. It is very likely that the complete, fully annotated version of the human genome sequence will be available before the initial deadline of 2005 and perhaps in time for the 50th anniversary of the discovery of the double-helical structure of DNA by Watson and Crick in 2003.
Strategy
DNA is sequenced in short reads of approximately 400 - 750 bp at a time. Smaller genomes such as those of bacteria and viruses have been sequenced in their entirety because of the limited challenge of piecing together “random” sequences. Larger genomes pose a special problem since randomly generated sequences (shotgun sequencing) would need “markers” to be able to assemble the complete sequence. This is analogous to a jigsaw puzzle - where the short, random sequences are the equivalent of pieces of the puzzle and the entire 3- billion bp sequence is the completed jigsaw puzzle. So to be able to piece together the entire picture, it was important to initially find “markers” across the entire genome that would subsequently allow the entire sequence of the genome to be assembled. The process of identifying these markers is referred to as the “mapping” of the genome.
The essential steps (Fig. 1) involved in facilitating the complete sequencing the human genome include the following: (a) genetic mapping, (b) physical mapping, (c) gene (transcript) mapping, and finally (d) shotgun sequencing.
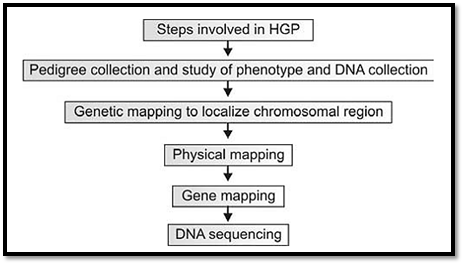
Fig. 1: Steps involved in human genome project
Variable sequences (i.e., polymorphisms), situated across the entire genome serve as genetic markers, and their identification constitutes the “genetic mapping” of the genome. Cutting up the genome into smaller segments, cloning them into vectors, and characterising the sequence of their ends to get small sequence tags (Sequence Tagged Sites/STS) across the genome constitutes the “physical mapping” of the genome. The vectors used for this purpose hold large DNA inserts (0.1 to 1 million bases). Using the STSs and other markers within the DNA inserts, clones spanning the entire genome were constructed and ordered.
Converting transcribed genes into their respective cDNAs by reverse transcription, and cloning and sequencing them was the basis of assembling a “gene or transcript map” of the genome. Using the latter method it was possible to sequence coding regions of a variety of unknown genes without knowing the corresponding genomic context or sequence. These sequenced genes also served the same function as STSs, and were called “Expressed Sequence Tags” (ESTs).
Having first mapped the genome using all these methods, it became possible to assemble randomly sequenced stretches of DNA into larger sequences (contigs) and subsequently to assembly the entire human genome. Genomic DNA was cut up into smaller bits using restriction enzymes and cloned into vectors that hold small inserts (2 - 50 kb) suitable for sequencing. This strategy is referred to as “shot-gun” sequencing, since essentially random bits of sequence are analysed from various clones. These sequences are then assembled by software programs using overlaps in sequence and the handles generated in the detailed mapping of the genome described above. Celera Genomics initially tried to assemble the entire genome using only sequence overlaps, but failed to do so and decided to also use the ordered markers generated by the international consortium and made freely available on the internet (GenBank).
The development of rapid and automated sequencing technologies, custom robotics, versatile sequence analysis and assembly software, and the over-whelming financial and logistical support of the public and private sectors helped bring about the spectacular success of the HGR.
SUMMARY OF THE HUMAN GENOME
polymorphisms): 1/1250 bp.
Medical benefits of the human genome project
Knowledge of the human genome sequence will have a profound influence in Medicine and Biology. Rather than the climax, the completion of the human genome sequence should be viewed as the beginning of the postgenomic era, a continued exploration of our molecular nature. From a medical standpoint, this knowledge will result in improved diagnosis and treatment of diseases with a genetic component.
Genes underlying inherited diseases are commonly identified by “positional cloning” i.e., using genetic linkage analysis in affected families, followed by gene hunting, and finally detection of mutations within the genes identified in the hunt. Before the advent of large-scale mapping and sequencing, this process typically took several years to accomplish. More recently, with the increasing availability of genomic sequence (released everyday over the past four years by the International Consortium), and the comprehensive mapping of the genome this process has become a lot less tedious. Consequently, at least 30 new disease genes have so far been identified that depended directly on the public release of the genomic sequence including those for breast cancer susceptibility (BRCA2), a form of limb girdle muscular dystrophy (LGMD2G), and three genes that cause non- syndromic deafness. The availability of the complete genome sequence makes it possible to comprehensively search for related genes. For example, after the identification of presenilin-1, a gene responsible for familial Alzheimer’s disease, a computer search of the genome identified a closely related gene (presenilin-2), which was also shown to cause autosomal dominant Alzheimer’s disease in other families.
The vast number of genes and their protein products are considered potential drug targets for the treatment of diseases. So far, a significant part of pharmaceutical research has been targeted towards the identification of such targets; however, the complete genome sequence has provided an instant treasure trove of potential targets for therapeutic strategies. Examples of these include (a) the identification of a previously unknown serotonin receptor, which is being investigated as a therapeutic target for schizophrenia, and (b) a new protease involved in the processing of beta-amyloid that is located on chromosome 21, which is being investigated as the cause of deposition of Alzheimer’s disease like amyloid in the brains of patients with Down’s syndrome.
With 99.999% of the genome being identical among all human beings, it is likely that the variable sequences (polymorphisms) in the genome may account for many of our individual differences. Single nucleotide polymorphisms (SNPs) occur once every 1250 bp in the human genome, and nearly 1.4 million have been identified to date. Less than 1% of these SNPs map within coding regions of genes and could potentially affect protein function. It is believed that “typing” of multiple SNPs will provide correlations with (a) susceptibility to certain disease states, especially those with a complex genetic contribution (e.g. type 2 diabetes, ischaemic heart disease, multiple sclerosis etc.), (b) differences in the response to drugs, and the incidence of side effects caused by them, and (c) disease prognosis.
Over 40% of the predicted genes have no ascribed function at this time. Their identification, without any relationship with a phenotype or function will result in accelerated research towards determining their role. Large- scale attempts to determine the function of these genes has resulted in the development of a new field called “functional genomics”. With the knowledge of the entire genetic complement of the human genome, we now know all the encoded proteins; the study of these using large-scale methods, for example in the comparison of “normal” versus “disease” tissues, is called “proteomics”. The availability of sequences from other organisms has also initiated the field of “comparative genomics” that is already having a major impact on the functional characterisation of the many newly identified genes.
Knowledge of one’s personal genetic information may be exploited by various agencies for many reasons. Insurance companies, the healthcare industry, employers, and government agencies may exploit this information. Genetic profiling could be a new basis for discrimination. It was recognized early on in the planning stages that the Human Genome Project would raise a multitude of ethical, social and legal issues. Already, the US and other European governments are in the process of drafting genetic privacy laws to prevent such information from being exploited. On the other hand, some societies do not seem to be overly concerned by this and have forged ahead forming unique relationships with the pharmaceutical industry. For example, the Icelandic government has developed collaborations with a pharmaceutical company, which will study their population for genetic variation and use it to develop new diagnostic, therapeutic and prognostic indicators.
Other genomes
Before the human nuclear genome was sequenced, genomic scientists had already sequenced genomes of the following: 599 viruses and viroids, 205 natural plasmids, 185 organelles (including the human mitochondrial genome in 1981), 31 eubacteria, 7 archaea, one fungus, two animals and one plant. With the technology in place, considerable effort is now being directed towards sequencing of other large genomes, such as the mouse, rat, zebra fish, puffer fish, and other primates. Plans are also being made to sequence other organisms that will help define key developments along the vertebrate and invertebrate lineages. Comparative genomics will then allow the characterization of evolutionarily conserved features and the identification of genetic innovations in specific lineages.
The future
The current draft version of the human genome sequence is expected to be fully completed by 2003. This however only represents the euchromatic portion of the genome; the heterochromatic portion is considered very difficult to sequence because it is largely composed of highly polymorphic tandem repeats. The details of all the genes and their splice forms remain to be unambiguously determined. Sequence similarity between human and mouse is likely to help identify over 95% of all exons and a significant proportion of the regulatory regions. This may not be too far off in the future since the mouse genome sequence will likely be completed within the next year. Plans are underway to clone every full- length human cDNA and have it available for the scientific community without restrictions.
Complete analysis of the polymorphic variation and its correlation with specific phenotypes holds great promise for the practice of medicine. Great advances will be made in the understanding of the molecular basis of disease which will likely lead to specific therapies. However, going from sequence to function will require a major concerted effort, perhaps larger in scale than the effort made in the Human Genome Project itself. These efforts will include the development of databases of gene expression, protein localization, protein-protein and DNA-protein interactions. New computational and technological advances will be required to fully realize the potential information embedded in the 3-billion bp of human DNA sequence now available.
References
Purandarey, H. (2009). Essentials of Human Genetics. Second Edition. Jaypee Brothers Medical Publishers (P) Ltd.

|
|
|
|
حمية العقل.. نظام صحي لإطالة شباب دماغك
|
|
|
|
|
|
|
إيرباص تكشف عن نموذج تجريبي من نصف طائرة ونصف هليكوبتر
|
|
|
|
|
|
قسم التطوير يختتم تدريب خمس مجموعات ضمن برنامج تمكين الخادم
|
|
|
|
المجمع العلمي يعلن إطلاق دوراته الصيفية في محافظة ذي قار
|
|
|
|
قسم شؤون المعارف يقيم ندوة حول القيمة العلمية والمعرفية للوثائق
|
|
|
|
قسم الإعلام ينظم دورة حول كتابة السيناريو
|