
آخر المواضيع المضافة

 النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية
النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية| BIOCHEMICAL GENETICS |


|
|
Read More
Date: 11-11-2015
Date: 10-11-2015
Date: 12-11-2015
|
BIOCHEMICAL GENETICS
INTRODUCTION
The way in which a mutation causes disease is largely related to deviations in the biochemical pathway. The path of molecular and biochemical events leading from a mutant gene to a disease gives us information not only about normal functioning but also about its deviation. Understanding these events can therefore help plan therapy for genetic diseases. Study of proteins and their metabolism constitutes the discipline of biochemical genetics.
The term “inborn error of metabolism” was established in 1902 by Sir Archibald Garrod when he observed that the urine of certain individuals turned black when exposed to air. Later it was observed that this was a genetic condition resulting from deficiency of a specific enzyme, causing a block in the normal metabolic pathway - in this case the protein being tyrosine. In another commonly known condition albinism, the deficiency of the enzyme tyrosinase in the hair, skin and eye prevents the synthesis of melanin leading to typical melanin deficient skin and irises. One has to understand that mutations producing biochemical defects do not always cause disease; they are biochemical traits detected in screening tests or when there is an offspring with a biochemical disease. In any population there is a normal variation in DNA sequences, which does not alter the quality of function of the polypeptide, irrespective of nucleotide changes being present in the coding region.
Genetic disease occurs when such alterations of vital genes reduce the quantity and function of the gene products (mRNA and protein). Single gene diseases occur due to alterations in DNA sequences controlling gene expression or encoding the structure of the protein.
In order to understand the pathogenesis of genetic disease, the primary biochemical abnormality must be understood. Till date 4,500 single gene disorders are known (autosomal and sex linked) and the specific protein deficiency has been identified in 500 such disorders. Inborn errors of metabolism are commonly caused by mutant genes, which generally result in abnormal proteins, most often enzymes. The inherited defects may be expressed as a total loss of enzyme activity or a partial deficiency of catalytic activity. Without treatment, the inherited defects often result in mental retardation or other developmental abnormalities due to harmful accumulation of metabolites. Some genetically determined enzyme deficiencies are discussed below (Fig. 1).
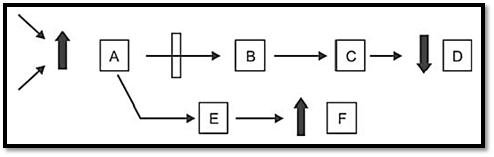
Fig. 1: Mechanism of genetic defect in metabolic pathway. Substrate A is converted to series of intermediate to final product D. Arrows indicate the enzymes catalyzing the reactions. A can converted to F by altering pathway. Genetic defect in the enzyme coverting A-B results in pathological consequences like 1. Accumulation of A, 2. Overflow of F, 3. Reduced formation of D, 4. Combination of these.
DISORDERS OF AMINO ACID METABOLISM
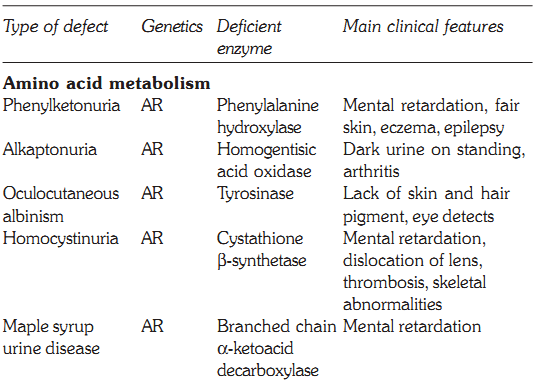
PKU is caused by a deficiency of phenylalanine hydroxylase, and is the most common clinically encountered inborn error of amino acid metabolism, with a prevalence of 1:11,000. Hyperphenylalaninemia may also be caused by a deficiency in the enzymes that synthesize or reduce the coenzyme tetrahydrobiopterin. Phenylalanine is present in elevated concentrations in tissues, plasma and urine. Phenyllactate, phenylacetate and phenylpyruvate are also raised. Clinically patients have mental retardation, failure to walk or talk, seizures, hyperactivity, tremor, microcephaly, and failure to grow. Virtually all untreated patients show an IQ below 50. Patients also show a deficiency of pigmentation (fair hair, light skin, and blue eyes). The high levels of phenylalanine present in PKU competitively inhibit the hydroxylation of tyrosine by tyrosinase, which is the first step in the formation of the pigment melanin. Classic PKU is caused by mutations in the gene that codes for phenylalanine hydroxylase (PAH) and is inherited in an autosomal recessive manner.
Regarding the treatment of PKU, blood phenylalanine is maintained by feeding synthetic amino acid preparations low in phenylalanine. The earlier the treatment is started, the more completely neurologic damage can be prevented. Treatment should not be delayed beyond the first month of life. Patients with PKU cannot synthesize tyrosine, and it should be supplemented in the diet.
Maternal PKU: When women with PKU who are not on a low phenylalanine diet become pregnant, the offspring are affected with maternal PKU syndrome. High blood levels in the mother cause microcephaly, mental retardation and congenital heart disease. Thus dietary control of phenylalanine must begin prior to conception.
Oculocutaneous Albinism (OCA)
This autosomal recessive disorder occurs due to deficiency of the enzyme tyrosinase, which is responsible for the formation of melanin pigment from tyrosine. OCA patients have lack of pigment in the skin, hair, iris and fundus of the eye (Fig. 2). The lack of pigment in the eye results in poor vision and nystagmus. OCA is a heterogeneous condition varying from tyrosine negative to tyrosinase positive form (measurable as tyrosinase positive and negative activity). DNA studies have shown that both these conditions occur due to mutations in the tyrosinase gene on the long arm of chromosome 11. Another condition called albinism type 2, OCA-2, has a mutation in the P gene located on chromosome 15.

Fig. 2: Oculocutaneous albinism
Alkaptonuria
Alkaptonuria was the first autosomal recessive inborn error of metabolism described by Garrod. It is caused by a deficiency in homogentisate oxidase. Homogentisic acid accumulates and is excreted in the urine. On exposure to air, it imparts a dark colour to the urine. The pigment also gets deposited in the wax of the ear, cartilage and joints. The condition is known as ochronosis and leads to arthritis in later life. Alkaptonuria is a benign condition and has a prevalence of 1 in 250,000.
Homocystinuria
This is inherited as an autosomal recessive disorder. The classical variety characterized by mental retardation, dislocation of the lens, convulsions, thromboembolic phenomena and osteoporosis. Arachnodactyly, kyphoscoliosis, and pectus excavatum may also be present.
The enzyme deficiency in homocystinuria is a lack of the enzyme cystathionine-B-synthetase. Accumulation of homocysteine occurs in the urine and methionine and its metabolites are elevated in blood. A positive nitroprusside test needs to be confirmed with plasma homocysteine levels. Treatment of homocystinuria is diet therapy, which involves low methionine, with supplements of cystine. Homocystinuria may be caused by decreased affinity of cystathione synthetase for its coenzyme pyridoxal phosphate (B6). This form may respond to megadoses of pyridoxine.
Disorders of branched chain AMINO ACID METABOLISM
The essential branched acids are leucine, isoleucine and valine. They share a common metabolic pathway in part and deficiency of this leads to maple syrup urine disease.
Maple Syrup Urine Disease
This is an autosomal recessive disorder and presents in the first week of life. Vomiting starts in the neonatal period and the neonates are alternately either hyper or hypotonic. If untreated, death is likely to occur in first few weeks. Neurologic problems are common and the prevalence of this disease is 1 in 200,000. The diagnosis is made initially by the typical smell of maple syrup in the urine. The deficiency of branched chain ketoacid dehydrogenase produces increased excretion in the urine of the branched chain amino acids valine, leucine and isoleucine. The confirmation is done by presence of these amino acids in urine and blood, and aminoacidograms by TLC and HPLC. The treatment is limiting dietary intake of these branched chain amino acids to the extent of the need of these essential amino acids for growth.
UREA CYCLE DISORDERS
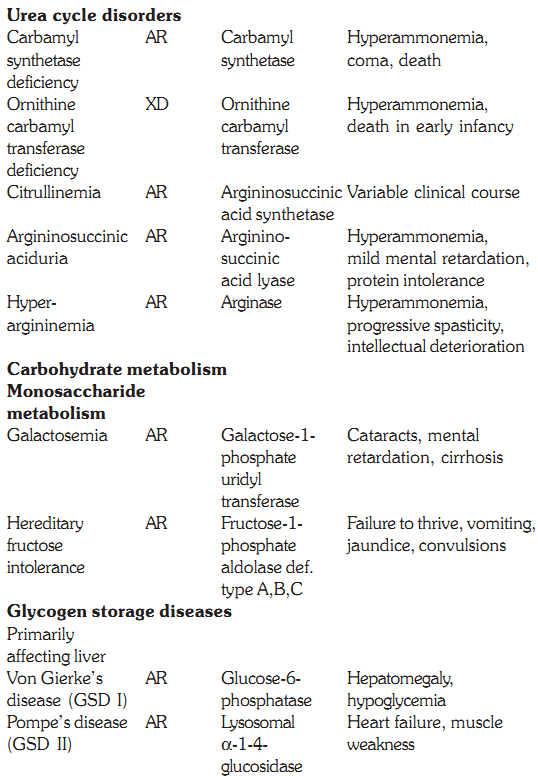
Urea cycle disorders are inherited as autosomal recessive disorders except for ornithine transcarbamylase deficiency, which is inherited as a X-linked disorder. The overall prevalence is 1 in 30,000 live births. The metabolic process of the urea cycle is a five step pathway, taking place primarily in the cells of the liver where waste nitrogen is removed from the amino groups of amino acids in normal protein turn over. In this reaction, two molecules of ammonia and one molecule of bicarbonate are converted into urea. The five enzyme deficiencies include carbamyl synthetase deficiency, ornithine carbamyl transferase deficiency, citrullinemia due to argininosuccinic acid synthetase deficiency, argininosuccinic aciduria due to argininosuccinic acid lyase deficiency and hyperargininaemia due to arginase deficiency. Deficiencies of enzymes in the cycle result in hyperammonemia due to intolerance of protein, and resulting mental retardation. High levels of ammonia are harmful to the central nervous system resulting in coma and death if untreated.
Disorders of carbohydrate metabolism
These include disorders of monosaccharide metabolism including galactosaemia, and hereditary fructose intolerance, and glycogen storage disorders.
Disorders of monosaccharide metabolism
Galactosemia
This is an autosomal recessive disorder, with a prevalence of 1:40,000 live births and occurs due to deficiency of the enzyme galactose-1-phosphate uridyl transferase, which is necessary for galactose metabolism. Untreated galactosemia presents with lethargy, feeding intolerance, vomiting hyperbilirubinemia, and liver dysfunction with coagulopathy. If not treated, 25% will develop sepsis in first 1-2 weeks. Mental retardation, cataracts and cirrhosis of the liver are the complications. Screening is by measurement of galactose and galactose-1 -phosphate, and confirmation is by measurement of the enzyme in erythrocytes. Early diagnosis can help in preventing complications, which also include speech abnormalities, behaviour problems, visual perceptual learning abnormalities and ovarian failure in affected females. The affected infants are treated with milk substitutes free of galactose and lactose, the common sugars found in milk (lactose is further broken down into galactose). Three other inborn errors of galactose metabolism are known, including variant forms of transferase deficiency, which occur with 10-35% of normal transferase activity, galactokinase deficiency, and uridinediphosphate-galactose-4-epimerase deficiency.
Hereditary Fructose Intolerance
This condition is inherited as an autosomal recessive manner occurring due to a deficiency of the enzyme fructose-1 phosphate aldolase. The normal source of fructose in the diet is honey, sucrose containing baby foods for example milk powder or cereals, fruits and certain vegetables. Fructose is also present in cane sugar with the disaccharide sucrose. Fructose intolerance can present at different ages. The symptoms are milder than those of galactosemia. It may present with hypoglycaemic coma, convulsions and death. The diagnosis can be confirmed by urine examination for fructose, by enzyme assays of the intestinal mucosa or by a liver biopsy sample. Restriction of fructose in the diet has a good long-term prognosis.
Glycogen storage diseases
These are a group of diseases that result from a defect in an enzyme required for either glycogen synthesis or degradation. They result in either formation of glycogen that has an abnormal structure or the accumulation of excessive amounts of normal glycogen in specific tissues. Due to block in the metabolic pathway the normal source of glucose is unavailable and this leads to hypoglycemia, liver function impairment and neurological abnormalities. There are six major types of glycogen storage disorders and there is one specific enzyme defect involving any one of the steps in the metabolic pathways of glycogen storage disorder. The glycogen storage diseases primarily affecting the liver are described first followed by those primarily affecting muscle.
Glycogen Storage Disease Affecting the Liver
von Gierke’s Disease (GSD I)
von Gierke’s disease is one of the first described disorder of glycogen metabolism is due to a deficiency of the enzyme glucose-6-phosphatase. This deficiency results in normal glycogen structure, but increased storage of glycogen. Affected infants have hepatomegaly, tachycardia due to severe fasting hypoglycemia, fatty liver, hyperlacticacidemia, and hyperuricemia. Treatment is offering frequent feeds and avoiding fasting.
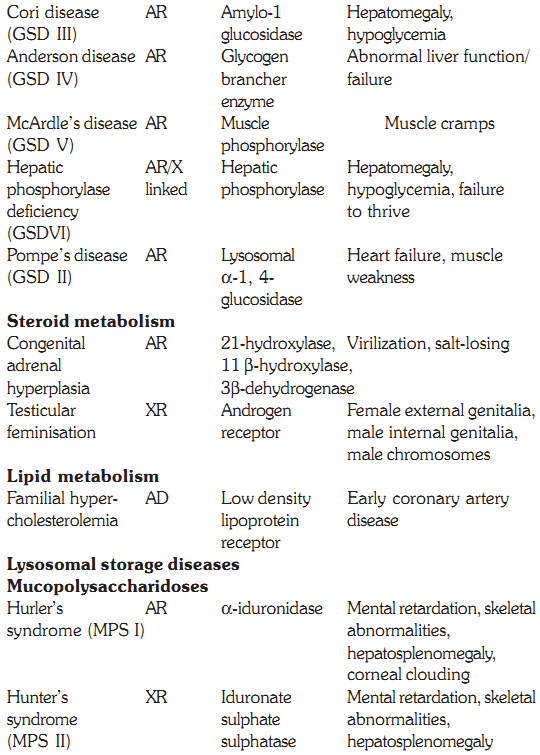
Cori Disease (GSD III)
Deficiency of the debrancher enzyme amylo-1-6 glucosidase results in accumulation of glycogen in the liver and other tissues. Affected infants present with hepatomegaly and muscle weakness. The treatment is the same as that for GSDI.
Anderson Disease (GSD IV)
Deficiency of glycogen brancher enzyme leads to Anderson disease. In this disorder there are long chains of glycogen, as very few branches are formed. Affected infants have abnormal liver function and hypotonia in first year of life, progressing rapidly to failure. There is no effective treatment except liver transplant.
Hepatic Phosphorylase Deficiency (GSD VI)
This multimeric enzyme complex is coded with subunits for both autosomal and X-linked genes. Deficiency of hepatic phosphorylase obstructs degradation of glycogen. Affected children present in the first 2 years with hepatomegaly, hypotonia and failure to thrive. Improvement in growth can occur with carbohydrate supplement.
Glycogen Storage Disease Affecting the Muscle
Pompe’s Disease (GSD II)
Infants with Pompe’s disease present in the first few months of life as floppy infants, with delayed motor milestones due to weakness of muscles. The heart is enlarged due to cardiac failure in the first or second year of life. Cardiac and skeletal muscles accumulate glycogen due to deficiency of lysosomal enzyme - 1, 4 glucosidase needed to break down glycogen. Excessive glycogen concentrations are found in abnormal vacuoles in the cytosol. The diagnosis is confirmed by enzyme assay on white blood cells or cultured fibroblasts.
McArdle’s Disease (GSD V)
These patients present in teenage years with complaints of muscle cramps on exercise. The symptoms appear due to deficiency of muscle phosphorylase, an enzyme necessary for degradation of muscle glycogen, the liver enzyme is normal. There is normal mental development and no rise in blood lactate during strenuous exercise. No effective treatment is known but in affected individuals muscle cramps can be reduced with continued exercise, the reason being utilization of other energy sources through other metabolic pathways.
Disorders of steroid metabolism
The products of adrenocortical steroidogenesis are glucocorticoids, mineralocorticoids and sex steroids. Inherited deficiency of various enzymes involved in cortisol and aldosterone synthesis leads to a group of diseases called congenital adrenal hyperplasia. The clinical characteristics of congenital adrenal hyperplasia (CAH) depend on which enzyme in the pathway of cortisol synthesis is deficient. Even for a specific enzyme, variability exists in the severity of disease expression and timing of onset of symptoms. The two most commonly seen defects are 21-hydroxylase deficiency and 11-hydroxylase deficiency.
Adrenogenital syndrome (congenital adrenal hyperplasia)
21- hydroxylase Deficiency
Accounts for 90% of cases and are inherited as autosomal recessive traits.
a- Classic salt wasting 21-hydroxylase deficiency: This is a severe deficiency resulting in decreased cortisol and aldosterone secretion, increased ACTH and 17 hydroxyprogesterone. Females infants are born with ambiguous genitalia, males have no genital abnormalities. Symptoms of salt wasting, vomiting, dehydration and shock develop in the first 2-4 weeks of life. Infants are hyponatremic, hyperkalemic, acidotic and often hypoglycemic.
b- Simple virilizing 21-hydroxylase deficiency: Clinical features are caused solely by overproduction of adrenal androgens. Therefore, only female infants with ambiguous genitalia are diagnosed during the neonatal period.
c- Non-classic 21-hydroxylase deficiency (acquired or late onset): This variant is diagnosed in female adolescents or adults and patients manifest signs and symptoms of androgen excess, like menstrual irregularities, hirsutism, acne and advanced bone age.
11-hydroxylase Deficiency
This accounts for about 5% of the cases of congenital adrenal hyperplasia. Patients manifest with hypertension and hypokalemia.
Abnormal sexual differentiation results in a newborn who appears sexually ambiguous, and can be classified as male pseudohermaphroditism, female pseudohermaphroditism, and abnormal gonadal differentiation.
Male Pseudohermaphroditism
Male pseudohermaphroditism refers to infants who are 46XY males and can be caused by a variety of endocrine disorders involving testosterone synthesis, metabolism or action at the cellular level.
1- Defects in testosterone synthesis and metabolism are caused by one of the 5 enzyme deficiencies inherited as autosomal recessive disorders. These include defects in cortisol synthesis and are classified as forms of CAH.
2- Defects in androgen action
a. 5α reductase deficiency impairs conversion of testosterone to dihydrotestosterone (DHT). Boys are born with ambiguous genitalia because DHT is necessary for masculinization of male external genitalia.
b. Androgen resistance syndromes (Testicular feminisation syndromes). In complete androgen resistance, an XY male infant with testes appears unambiguously female because of complete resistance to androgen action at the cellular level. In partial androgen resistance, the affected XY individual has ambiguous genitalia.
Female Pseudohermaphroditism
Female pseudohermaphroditism refers to infants who are 46XX females with ovaries, who appear masculinized at birth. This can be caused by CAH (discussed above) or maternal androgen or progestin exposure.
Abnormal Gonadal Differentiation
True hermaphroditism occurs when there is both ovarian and testicular tissue in the gonads. In 80% of the cases the karyotype is 46XX. Mixed gonadal dysgenesis has a karyotype of 45X/ 46XY. There is a spectrum in the appearance of the genitalia from completely male to completely female.
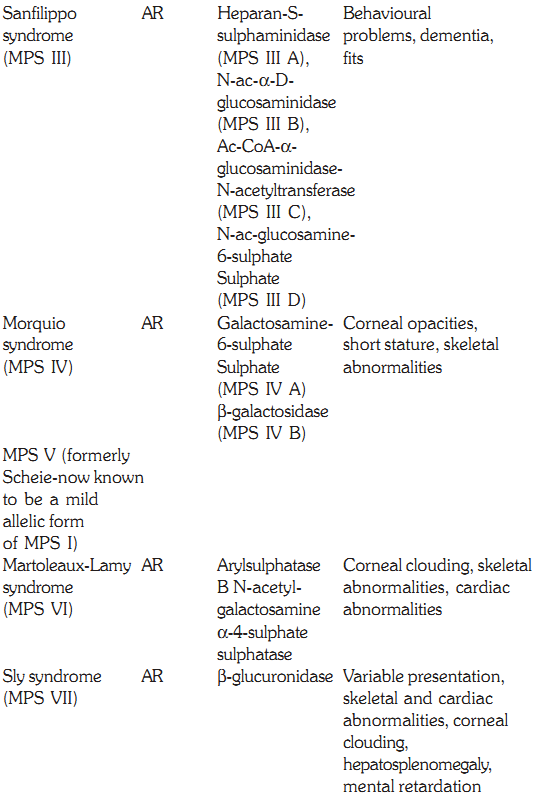
MUCOPOLYSACCHARIDOSES
These are hereditary disorders that are clinically progressive and are characterized by accumulation of glycosaminoglycans in various tissues causing skeletal and extracellular matrix deformities. Mucopolysaccharidoses are caused by a deficiency of one of the lysosomal hydrolases normally involved in the degradation of one or more of the glycosaminoglycans. They also result in oligosaccharides in urine due to incomplete degradation of glycosaminoglycans. All the deficiencies are autosomal recessive except Hunters syndrome, which is X-linked. No effective therapy exists, but prenatal diagnosis for these deficiencies is possible by measurement of lysosomal hydrolases.
Hurler’s Syndrome (MPS I)
Hurler’s syndrome is the commonest type of MPS and is clinically more severe. The symptoms present in the first year of life with corneal clouding, curving of lower thoracic and lumbar spine, and poor growth. Patients develop hearing deficiency and enlarged liver and spleen. During the second year of life, vertebral changes with stiffening of the joints occur. The facies is characteristically coarse. The features start progressing along with mental deterioration, and death occurs in mid teens due to respiratory infections and cardiac failure. Hurler syndrome is diagnosed by a urine test showing excretion of dermatan and heparan sulphate. Confirmation is done by demonstration of reduced activity of the α-L iduronidase lysosomal hydrolase. Levels of residual α-L iduroindase activities vary and on this basis the disease was separately classified as Scheie’s disease (MPS IS) and Hurler / Scheie disease (MPS I H/S), which are allelic disorders.
Hunter’s Syndrome (MPS II)
This is inherited as an X-linked disorder and affected males present between 2-5 years of age with hearing loss, diarrhea, recurrent infections and poor growth. Clinical examination reveals coarse features (Fig. 2), enlargement of liver and spleen and stiffness of joints. X-rays of the spine show lipping of the vertebral bodies. Both physical and mental retardation are progressive and death occurs before the age of twenty. Confirmation of MPS is by a urine test and further classification by an assay of the enzyme iduronate sulfatase sulphatase in serum or WBCs.

Fig. 2: Hunter’s syndrome (MPS II)
Sanfilippo Syndrome (MPS III)
This form of MPS is seen commonly. The disease presents itself in the second year of life. The facial features become coarse, and skeletal changes and there is progressive intellectual loss. There may be associated behavioural problems. Convulsions occur oten and death results in the twenties or thirties. Four enzymatic steps are necessary and the deficiencies are described below. Type A has heparan sulphaminadase deficiency, Type B has N-acetyl glycosaminadase deficiency, Type C has N-acetyltransferase deficiency and Type D has N-acetyl glucoseamine deficiency. Though the enzymes involved are different, clinical presentation remains the same.
Morquio Syndrome (MPS IV)
Morquio syndrome (MPS IV) is seen in the second or third year of life with skeletal abnormalities. These include short stature, thoracic deformity and kyphoscoliosis. Patients have a normal IQ and the prognosis for long-term survival is good, though there is an increased risk of spinal cord compression. Urinary test is used for screening and confirmation by enzyme assays shows a deficiency of N-acetylgalactosomine sulphate (MPS IV A) or B Galactosidase (MPS IV B)
Martoleaux-Lamy Syndrome (MPS VI)
Patients with Martoleaux-Lamy syndrome (MPS VI) have an expression similar to that of Hurler syndrome. The intelligence is normal. Life expectancy is up to early adulthood but in minor forms, patients may live up to the third decade. The diagnosis is confirmed by deficiency of arylsulphatase B deficiency in fibroblasts and WBCs.
Sly Syndrome (MPS VII)
This syndrome has variability in the severity and involvement of the systems, ranging from mild kyphoscoliosis to coarse facial features. It is caused by a deficiency of b glucuronidase. Corneal clouding, cardiac anomalies, hepatosplenomegaly, and a low IQ are other features, and death occurs in early childhood. There is an increase in urinary glyosaminoglycans. Enzyme assays in white blood cells or in fibroblasts confirms the diagnosis. Treatment by enzyme replacement has been unsuccessful so far. However, bone marrow transplant has met with limited success.
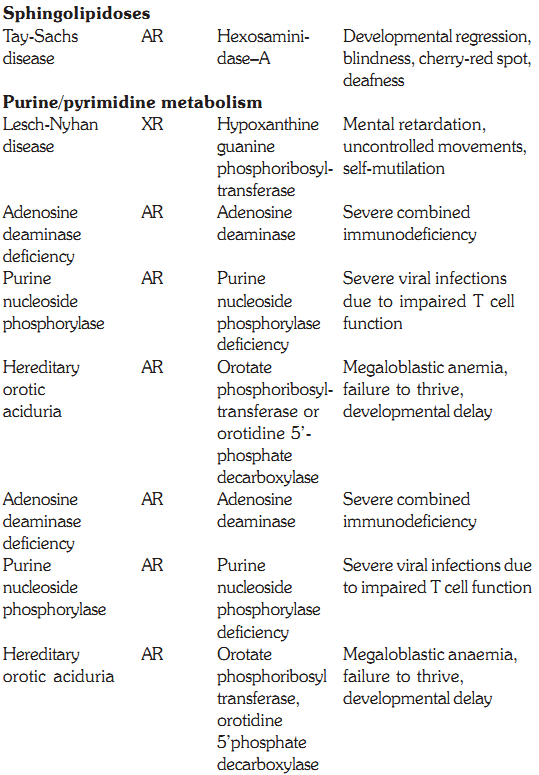
SPHINGOLIPIDOSES
In a normal individual, the synthesis and degradation of sphingolipids are balanced, so the amount of the compounds present in the membranes is constant. If a specific hydrolase required for the degradation process is partially or totally missing, sphingolipids accumulate in the lysosomes and are called sphingolipidoses. These include Tay Sach’s disease, GM1 gangliosidosis, Gaucher’s disease, metachromatic leukodystrophy, Krabbe’s disease, Sandhoff’s disease, Fabry’s disease, Niemann Pick disease and Farber’s disease. All of the above are autosomal recessive diseases except for Fabry’s disease, which is X-linked. The incidence of sphingolipidoses is low in most populations except for Gaucher’s disease and Tay Sachs disease, which show a high frequency in Ashkenazi Jews. The diagnosis of sphingolipidoses can be made by the presence of enzyme activity and accumulated lipid by analysis of tissue samples, cultured fibroblasts, peripheral leukocytes, plasma and amnio tic fluid (for antenatal diagnosis).
Tay-Sachs Disease
It occurs due to increased gangliosides due to a deficiency of b hexosaminidase A. This disorder occurs commonly in
Ashkenazi Jews with an incidence of 1 in 2600 individuals. Affected infants start showing symptoms within a few months after birth with poor feeding, lethargy and floppiness. Developmental regression continues and becomes apparent in the later half of the year. Visual impairment and deafness occur and spasticity increases with rigidity in limbs. Death occurs by the age of 3 years. There have been reports of juvenile and adult forms. Presence of a cherry red spot in the centre of the macula confirms the clinical diagnosis of Tay-Sachs disease. Laboratory confirmation is done by demonstrating reduced hexosaminidase levels in serum, WBCs or cultured fibroblasts.
Gaucher’s Disease
It occurs due to increased glucocerebrosides due to a deficiency of b glucosidase. Gaucher’s disease is a common type of sphingolipidoses seen in Ashkenazi Jews. The age of onset differs in patients of Gaucher’s disease, and based on this they are divided in 2 groups. The adult type or type I is more common and affected persons present with symptoms of with pain in limbs and joints and a tendency to pathologic fractures. Clinically patients are anemic with an enlarged spleen and liver. There are bony changes seen on X-rays in the vertebral bodies and femora. In infantile Gaucher’s disease or type II, there is involvement of the central nervous system. The age of onset is 3-6 months. Failure to thrive, neurological deterioration and developmental regression occur together with convulsions and increasing spasticity. Death occurs in the second year of life due to recurrent respiratory infections. The diagnosis is confirmed by reduced levels of P-glucosidase in WBCs and cultured fibroblasts.
Treatment for adult type of Gaucher’s disease is symptomatic. Splenectomy is occasionally required which can however cause secondary anemia. The current treatment with enzymes is by modifying P-glucosidase by addition of mannose- 6-phosphate. Dramatic improvement in symptoms and regression of spleen is noted.
Niemann-Pick Disease
It occurs due to increased sphingomyelins due to a deficiency of sphingomyelinase. Clinically patients with Niemann-Pick disease present in infancy with failure to thrive, hepatomegaly, developmental regression and presence of a cherry red spot in the macula (also occurs in Tay-Sach’s disease). Death occurs by the age of 4 years. Diagnosis is confirmed by presence of deficiency of the enzyme sphingomyelinase. Bone marrow of patients with Niemann-Pick disease show typical foam cells due to sphingomyelin accumulation.
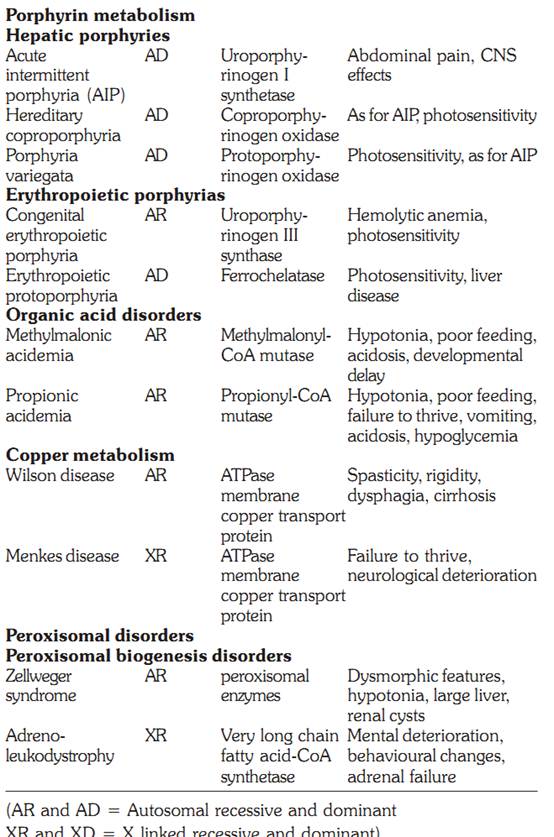
Copper metabolism
Copper homeostasis is maintained by gastrointestinal absorption and biliary excretion. Inherited disorders of copper transport include Wilson’s disease, Menkes disease, and aceruloplasminemia.
Wilson Disease
The Wilson disease copper transporting adenosine triphosphatase (ATPase) transports copper into the hepatocyte secretory pathway for incorporation into ceruloplasmin and excretion into the bile. Thus individuals present with signs and symptoms arising from impaired biliary copper excretion. The disorder is inherited in an autosomal recessive manner, and a large number of mutations have been identified in the Wilson disease gene. The impaired copper excretion results in accumulation in the liver. When the capacity for hepatic storage is exceeded, the copper is released into the plasma resulting in haemolysis and deposition of copper in extra-hepatic tissues. Affected individuals may present with chronic hepatitis and cirrhosis or acute liver failure. Copper deposition in the retina is called the Kayser Fleischer ring. Copper accumulation in the basal ganglia and other parts of the brain results in dystonia, tremor, personality changes and cognitive impairment. The diagnosis is confirmed by decreased serum ceruloplasmin, increased urinary copper, and elevated hepatic copper concentration. The treatment is copper chelation using penicillamine.
Menkes Disease
The Menkes disease ATPAse transports copper across the placenta, gastrointestinal tract and blood brain barrier and clinical features of this disorder result from copper deficiency. In the fetus, there is copper deficiency in utero. The clinical features include abnormal hair and pigmentation, laxity of the skin, metaphyseal dysplasia, cerebellar degeneration and failure to thrive. Decreased serum copper and ceruloplasmin confirm the diagnosis. The disorder is inherited in an X-linked manner and mutations found in the gene for Menkes disease are unique to each family.
Aceruloplasminemia
This is an autosomal recessive disorder characterized be absent serum ceruloplasmin due to mutations in the ceruloplasmin gene. It is characterized by progressive neurodegeneration due to iron accumulation in the basal ganglia. Thus ceruloplasmin has an essential role in iron homeostasis. Affected individuals present with dysarthria, dystonia and dementia due to iron accumulation in affected tissues.
PEROXISOMAL DISORDERS
Peroxisomes are single membrane lined organelles present in virtually all eukaryotic cells and range from 100-1000 peroxisomes per cell. Peroxisome biogenesis involves synthesis of the matrix proteins on free cytosolic ribosomes followed by receptor mediated import into the organelle. PEX genes encode peroxins, proteins involved in and necessary for peroxisome biogenesis. There are 15 PEX genes known in humans. The peroxisome disorders are comprised of at least 12 complementation groups. Defective biogenesis of the organelle leads to two clinical spectra: The Zellweger spectrum, which includes Zellweger syndrome, neonatal adrenoleukodystrophy and infantile Refsum disease. The second spectrum consists of rhizomelic chondrodysplasia punctata. The gene for X-linked adrenoleukodystrophy also codes for a peroxisomal membrane protein, and hence this disorder will also be discussed.
Zellweger Syndrome
This cerebrohepatorenal syndrome has an incidence of approximately 1 per 50,000 births, and affected infants rarely live more than a few months. Patients have multiple congenital anomalies as well as ongoing metabolic disturbances. There are characteristic features including large anterior fontanel, full forehead, hypoplastic supraorbital ridges, epicanthal folds, broad nasal bridge, and a small nose with anteverted nares, cataracts, glaucoma, corneal clouding, Brushfield spots, pigmentary retinopathy, and optic nerve dysplasia, severe hypotonia, weakness and neonatal seizures. Radiologic examination reveals abnormal punctate calcifications (calcific stippling) in the patella and epiphyses of long bones. The cause of Zellweger syndrome is failure to import newly synthesized peroxisomal proteins into peroxisomes. There is plasma and tissue accumulation of very long chain fatty acids (VLCFAs).
Infantile Refsum Disease
Infantile form of Refsum disease can be caused by mutations in the PEX1 and PEX2 genes. Patients with the infantile form show both clinical and biochemical differences from patients with the classic form of Refsum disease. Features include early onset, mental retardation, facial dysmorphism, sensorineural hearing loss, hepatomegaly, osteoporosis, failure to thrive, delayed development, mental retardation, hepatomegaly, skeletal changes and retinitis pigmentosa. The biochemical abnormalities include accumulation of very long chain fatty acids (VLCFA). Deficiency of peroxisomes in hepatocytes and cultured skin fibroblasts is demonstrable. Biochemically, IRD patients show accumulation of phytanic acid and defective bile acid metabolism.
Rhizomelic Chondrodysplasia Punctata (RCDP)
RCDP is a rare, multisystem, developmental disorder, characterized by the presence of stippled foci of calcification in hyaline cartilage, coronal vertebral clefting, dwarfing, joint contractures, congenital cataract, ichthyosis, and severe mental retardation. Biochemically, RCDP patients have subnormal levels of red cell plasmalogens and progressive accumulation of phytanic acid starting from normal at birth and increasing to levels more than 10 times normal by age 1 year.
Adrenoleukodystrophy
Two types of adrenoleukodystrophy are known, X-linked adrenoleukodystrophy (X-ALD), and autosomal recessive neonatal adrenoleukodystrophy, which resembles Zellweger syndrome. The gene for X-linked ALD maps to Xq28 and encodes a peroxisomal membrane protein with homology to the ATP-binding cassette (ABC) transporter family of proteins. The incidence of males with X-ALD is between 1-20,000 to 50,000. The disorder causes malfunction of the adrenal cortex and nervous system myelin, and is characterized by abnormally high levels of very long chain fatty acids in tissues and body fluids. The very long chain fatty acid (VLCFA) accumulation is associated with an impaired capacity for their degradation, a reaction that takes place in the peroxisome. Clinically the children with the childhood cerebral form suffer from dementia, adrenal insufficiency and progressive neurologic deficit. Bone marrow transplantation is the most effective therapy in children who show early evidence of cerebral involvement. Dietary therapy to reduce plasma VLCFA levels is using a 4:1 mixture of glyceryl trioleate and glyceryl trierucate known as Lorenzo’s Oil.
Disorders of purine and pyrimidine metabolism
Defects in the Purine Salvage Pathway
Purines that result from the normal turnover of cellular nucleic acids or those that are obtained from the diet and are not degraded can be reconverted into nucleoside triphosphates and used by the body. This is the salvage pathway for purines, and two enzymes are involved, APRT (adenine phosphoribosyl transferase) and HGPRT (hypoxanthine guanine phosphoribosyl transferase). A deficiency of HGPRT causes Lesch Nyhan syndrome, discussed below.
Lesch Nyhan Syndrome
Defects in the degradation of purine nucleotides
Purine nucleotides are sequentially degraded to form uric acid. Several genetic diseases are associated with deficiencies of specific degradative enzymes in this pathway. Some of these are discussed below.
a- A defect in adenosine deaminase (ADA) deficiency causes severe combined immunodeficiency (SCID) involving T-cell and B-cell dysfunction. dATP is the major nucleotide that accumulates in cells.
b- Purine nucleoside phosphorylase deficiency results in impairment of T-cell function but no apparent effect on B-cell function. This results in decreased uric acid formation combined with increased levels of purine nucleosides and nucleotides. dGTP is the major nucleotide that accumulates in red cells.
Gout
Gout is characterized by hyperuricemia with recurrent attacks of acute arthritic joint inflammation, caused by deposition of uric acid crystals. Primary gout is attributable to an inborn error of metabolism such as overproduction of uric acid. Treatment with allopurinol inhibits xanthine oxidase resulting in the accumulation of hypoxanthine and xanthine, which are compounds that are more soluble than uric acid.
Hereditary Orotic Aciduria
This condition presents in children during first year of life. The children fail to thrive and have megaloblastic anemia and delayed development. The condition is due to deficiency of enzymes orotate phosphribosyl transferase or orotidine-5’ phosphate decarboxylase. These enzymes are important in pyrimidine synthesis. Large amounts of orotic acid are excreted in the urine. Therapy is with uridine, which reduces orotic acid excretion and is aimed at correcting growth and anemia.
Prenatal diagnosis of inborn errors of metabolism
The first criterion in the prenatal diagnosis of an IEM is to have a confirmed laboratory diagnosis in an index case in whom the deficient gene product has been identified. First trimester prenatal diagnosis is possible by cultured chorionic villi and it is not necessary to wait until second trimester for amniotic fluid culture. With the use of recombinant technology, it is possible to use linked DNA sequences or mutations and this is of great value when the biochemical basis has not been established or the enzyme in question is not expressed in chorionic villi.
Some defects, their genetics, deficient enzymes and clinical features are discussed below:
Characteristics of some inborn errors of metabolism (AR and AD = autosomal recessive or dominant. XR and XD = X-linked recessive or dominant
References
Purandarey , H. (2009) . Essentials of Human Genetics. Second Edition. Jaypee Brothers Medical Publishers (P) Ltd.

|
|
|
|
تفوقت في الاختبار على الجميع.. فاكهة "خارقة" في عالم التغذية
|
|
|
|
|
|
|
أمين عام أوبك: النفط الخام والغاز الطبيعي "هبة من الله"
|
|
|
|
|
|
|
قسم شؤون المعارف ينظم دورة عن آليات عمل الفهارس الفنية للموسوعات والكتب لملاكاته
|
|
|
















