
آخر المواضيع المضافة

 النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية
النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية| Aligning Sequences |


|
|
Read More
Date: 7-12-2015
Date: 9-12-2015
Date: 29-12-2015
|
Aligning Sequences
The most basic activity in sequence analysis involves aligning protein or nucleotide sequences together. This need arises due to the processes of molecular evolution: gene duplication followed by continual divergence of the sequences through the accumulation of mutations over time. Comparative biological analysis, which has long been such a powerful tool for biologists (as exemplified by Linnaeus and Darwin), is arguably even more applicable in sequence analysis than in any other branch of biology, because it can be applied to an enormous number of character states at the level of individual residues in nucleic acid or protein sequences. First, however, related sequences must be correctly aligned before the power of comparative analysis can be brought to bear. Because of the difficulty of aligning highly diverged sequences, and the many applications of sequence alignment, this is one of the most active areas for method development in computational biology.
Alignment tasks generally divide into pairwise sequence alignment and multiple sequence alignment, although the underlying algorithms may share many details. The most sensitive methods for aligning sequences belong to the class of algorithms known as dynamic programming (or minimum string edit) that were initially developed for applications in text comparison. Two of the dynamic programming algorithms most used in biology are usually known as Needleman–Wunsch (1) and Smith–Waterman (2), after the researchers who first applied them to biological sequences. Because these algorithms allow gaps to be inserted at any position in the sequences, they are computationally slow. By contrast, word comparison algorithms, which do not allow gaps, are much faster, but at the expense of accuracy and sensitivity in aligning sequences.
1. Pairwise Sequence Alignment Algorithms
1.1. Dynamic Programming
The basic algorithm works through a two-dimensional matrix in which every residue in one sequence is scored against every residue in the other sequence (1). The algorithm begins in one corner (eg, top left) of the matrix and ends in the opposite corner (bottom right). At each point in the matrix, the algorithm iterates the same set of choices. Typically, it chooses which of three existing paths scores best when extended into the current point: (i) match the residues and continue aligning from the previously aligned residue pair, or (ii) pay the penalty and insert a one-residue gap into sequence X, or (iii) pay the penalty and insert a one-residue gap into sequence Y (see Gap Penalty). The algorithm is guaranteed to find the best path through the matrix, allowing for gaps at any position in either sequence. Scores for matching the residues are taken from residue exchange, or mutation, matrices. For nucleotide sequences, these are usually quite simple: for example, +1 for an identity, 0 for a transition, and –1 for a transversion. For proteins, typical exchange matrices are the more complex 20 * 20 PAM (point accepted mutation) matrices introduced by Margaret Dayhoff (3), or subsequently derived PAM series derived from larger alignment datasets (4, 5). A PAM 250 matrix is shown in Figure 1. Gap penalties are used to control the frequency and length of gaps inserted in the sequences. Where appropriate, varying gap penalties in a position-specific manner can improve the alignment.
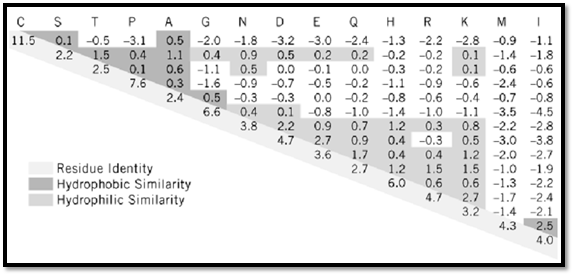
Figure 1. PAM250 amino acid exchange matrix developed by Gonnet and colleagues (4) and superseding the original D acids have positive log-odds exchange values while dissimilar pairs have negative values. All positive scores are coloredpurple highlights similar pairs of hydrophobic residues, and green indicates similar pairs of hydrophilic residues. The hig aromatic residues (Phe, Tyr, Trp) and are stronger than exact matches of highly mutable residues such as Ser. The strong hydrophobic residues and small or negatively charged residues.
Dynamic programming algorithms work through a two-dimensional matrix of area M*N in aligning sequences of lengths M and N. Therefore the computational requirement [usually symbolized as O) MN)] has a constant factor for the calculation, multiplied by the two sequence lengths. To obtain an alignment, the algorithm makes a first pass to determine the end of the highest scoring matched segment and then a second pass working back to obtain the alignment. If only the highest score, but not the actual path, is needed (as in a database search), then only the first pass need be done. Naive implementations that first plot the whole matrix to an array will also use O(MN) memory. Because the algorithm works through the array systematically, however, it is unnecessary to store the whole array in memory. Memory-efficient implementations of the first pass are straightforward, because the alignment is not being kept (6). The second pass, to obtain the alignment, is more complicated, but memory-efficient recursive methods have been developed that have allowed large alignment tasks to be ported to small personal computers, with a small but acceptable loss in calculation speed (7-9) .
1.2. Global Alignment
The standard Needleman–Wunsch algorithm (1) finds the optimal full-length alignment for a pair of sequences. Global alignment is appropriate where sequences are known to be both homologous and collinear and is therefore often used for multiple alignment of sequence families.
1.3. Best Local Alignment
Variants of the Smith–Waterman algorithm (2) find the optimal alignment that has a positive value for the path for a given pair of sequences. Except for highly related sequences, the best local alignment is a partial match between the sequences. Residue exchanging mutation matrices, such as PAM250, provide log-odds scores for the likelihood that a pair of residues will exchange as a result of mutation: Similar residues that exchange easily have positive log scores, while dissimilar residues have negative log scores. The best local alignment is taken as the highest-scoring continuously positive path. This algorithm is appropriate under conditions where sequences are not known to be both fully homologous and collinear—for example, multidomain proteins, or DNA regions containing rearrangements. Smith–Waterman type algorithms underlie the most sensitive methods for database searching by sequence homology yet to be devised. Because of the computational cost, they are not applied as often as search methods using ungapped alignment algorithms.
The Waterman–Eggert (10) extension of the algorithm will return sets of suboptimal paths that do not intersect with the optimal path, and in this way it can find repeats in sequences.
1.4. Best Local Ungapped Alignment
Widely used database search tools, such as BLAST (11) and FASTA (12), search for the highest scoring matched regions without allowing for gaps. Word searches and other ungapped alignment methods are much faster than dynamic programming approaches, but at the expense of sensitivity. Thus these methods are likely to miss homologous but divergent matches. To improve the results, FASTA does a second dynamic pass on the set of top hits. For a small reduction in search speed, BLAST2 examines the gap cost between the set of ungapped positive matches between two sequences and returns composite best locally aligned regions, including gaps whenever the score is still positive. The latter algorithm is likely to approach Smith–Waterman sensitivity except for the most unusual alignment circumstances.
2. Multiple-Sequence Alignment
This is a set of homologous protein or nucleotide sequences that have been correctly aligned, allowing for the presence of indels. Figure 2 shows an aligned region for some elongation factor TU sequences.
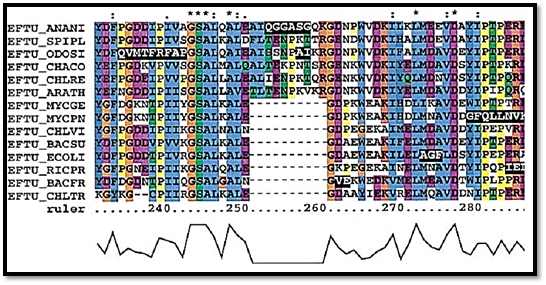
Figure 2 : aligned region for some elongation factor TU sequences
2.1. Uses of Multiple Alignments
Multiple alignments are indispensable in computational biology. They are the basic dataset used to construct phylogenetic trees, which are themselves important computational tools (eg, for weighting sequences by divergence) as well as providing insight into past evolution. They reveal conserved residues that are likely to be structurally or functionally (eg, in catalysis) important and unconserved positions that either are unimportant or have acquired a change of function (Fig. 2). They improve the accuracy of many sequence analysis functions as compared to single sequences, such as secondary structure prediction (13), coiled-coil prediction (14), and transmembrane helix prediction (15). They are useful as the query input for the most sensitive homology searches (alignment profile (16) and hidden Markov model searches (17)) and can be used to detect divergent homologues that single-sequence queries cannot pick up. They are useful in the detection of domains and in defining their boundaries in modular, mosaic proteins (18). Multiple alignments of folded RNAs are a prime resource used in determining their secondary and tertiary structures, by mapping residue conservation and long-distance-coupled mutations (19, 20). DNA multiple alignments are used for identifying conserved signals, such as promoter elements and RNA splice sites (21).
2.2. Multiple-Alignment Algorithms
So far it has been necessary to adopt heuristic strategies to generate multiple alignments, because formally correct methods have been computationally impractical to implement. The ideal method to align N sequences would be N-dimensional dynamic programming, as this would be guaranteed to find the optimal path (ie, the optimal multiple alignment) in an N-dimensional matrix. Unfortunately the computer time required to align N sequences of length l is O(lN) and is impractical for more than three or four sequences, although by limiting the search space to likely regions, the MSA program can align up to eight sequences (22). Another broad class of methods, those that iterate toward an optimized score for the alignment, are still computationally intensive but are becoming practical with increasing computer power. These include a number of approaches, some of which may be used in combination, such as global minimization, genetic algorithms, and trained neural networks (23, 24). The goal is to harness a good model description of a multiple-sequence alignment with an effective iteration strategy, so as to get high-quality alignments in a practical timescale. In the meantime, widely used alignment programs such as Clustal W (25) follow the heuristical clustered alignment strategy.
2.3. Progressive Clustered Alignment
This two-step approach was introduced by Feng and Doolittle (26) in an attempt to minimize errors in the final multiple alignment, by aligning the most similar sequences first and the most divergent ones last. The set of unaligned sequences are first aligned in pairwise fashion to each other, so that a matrix of the approximate pairwise similarities may be obtained. A matrix-based tree construction method, such as Neighbor-Joining (27), is used to construct a dendrogram linking the sequences according to their observed similarities. Guided by the dendrogram branching order, the sequences are then sequentially aligned together using dynamic programming, beginning with the most closely related sequences and ending by merging the most divergent groups by profile alignment. This procedure minimizes alignment errors, which become more likely with increasing sequence divergence, becoming a problem for proteins less than 25% to 30% identical in sequence. The final alignment should always be examined for misalignment, especially of the more divergent sequences, because such errors are likely to be present in any but the most straightforward multiple alignment task.
The sensitivity of the basic clustered alignment strategy can be improved by several modifications, such as weighting the sequences by divergence and position-specific gap penalties (25). Where tertiary structure information is available, gap penalty masks can be employed to guide the gaps into regions of sequence that are expected to be tolerant of indels (28).
2.4. Profile Alignment
A set of aligned sequences can be aligned to one or more new sequences by treating the group much as a single sequence. The score for one alignment column can be obtained by summing the log-odds residue exchange scores for the observed set of amino acids (16), correcting for sequence relatedness by downweighting similar sequences (25). Conserved alignment columns score more highly than unconserved columns, where the log-odds scores tend to cancel each other. Gap penalties can be lowered at existing indels, because gaps in new sequences are more likely at these positions than at ungapped positions. The improvement in signal to noise provided by the extra information in the alignment means that a profile alignment is more accurate than independent pairwise alignment of the same set of sequences. As well as being used to merge aligned groups in the clustered alignment strategy, profiles provide a sensitive search strategy to find highly divergent homologues and are one of the main sequence analysis tools for identifying protein domain families (18).
2.5. Hidden Markov Model (HMM) Alignment
HMMs are a class of probabilistic models, applicable when the components of a complex linked system behave independently (a so-called Markov chain). Up to a point, this is valid for residue mutations in globular protein sequences, and HMMs are being applied increasingly in multiple-alignment algorithms and profile-type database searches (17, 29). The models are more complex than the widely used PAM model for protein evolution introduced by Margaret Dayhoff (3), providing both advantages and disadvantages. On the plus side, the models provide direct
probabilities for evaluating database search matches, which can include multiple matches in a repeated sequence and can formally treat biological complexities such as splice junctions in genomic sequence. On the debit side, the extra parameters lead to more complex optimization problems at several levels. Thus, inexperienced users may set up poorly optimized HMMs while, for program developers, there is a problem as to whether the most appropriate HMMs are being applied to sequence evolution. It is important for the user not to be seduced by technical jargon, but to justify the use of newer methods such as these on the basis of convincing results.
3. Error in Sequence Alignments
Errors are very common and invalidate any conclusions obtained by methods based on sequence alignments (see Fig. 2). The three main sources of error are: the input sequences, mistakes by the user, or alignment algorithm failure. Causes and effects of error are manifold; some major ones are discussed below.
3.1. Errors in the Input Sequences
Experimental errors in determining sequences include double-insert cloning errors, truncated cDNA clones, base insertion or deletion causing translation frameshifts, and translation with inappropriate genetic code (eg, for plastid-encoded proteins). Figure 3 shows an example of a frameshift error in a database entry. Algorithm failure during alignment may be induced by sequence error, such as frameshifted regions, in which the sequences are no longer similar, and may induce incorrect gap placement (Fig. 2). Errors in sequences are very common and likely to be present in any sequence family (30, 31).

Figure 3. Frameshifted segment in the Mycoplasma pneumoniae EFTU sequence revealed by comparing the DNA to the closely related Mycoplasma genitalium EFTU using a dynamic programming three-frame comparison allowing shifts bet translation frames (30). Exclamation points mark the frameshift sites. The first frameshift is caused by a base being dropp and the second frameshift is caused by a base being added, thereby returning to the original translation frame. See Figure multiple alignment spanning this region.
Further errors can arise in preparing sequence database entries. These can affect any part of the entry and can have particularly unpredictable consequences. However, the most common annotation errors are undoubtedly in predicted translation products and are a consequence of the limited accuracy of current gene prediction algorithms; that is, despite high-quality DNA sequence, the predicted protein sequence might contain artificially translated introns, missed exons, or terminal truncations or might be an artificial fusion product of two independent genes. Protein sequence alignments can often help in identifying translation problems.
3.2. Mistakes by the User
Generally these are due to inadequate attention to detail. Erroneous inclusion of nonhomologous sequences in the input set is quite common, particularly if keyword searches are used to extract a set of sequences; there are many examples of unrelated proteins performing equivalent functions, as well as sequences that have functions incorrectly ascribed to them. Another source of error occurs during the alignment process, when parameters may be set poorly. Trial and error is usually needed to find the parameters that best suit a particular group of sequences. For example, insertion of too few or too many gaps may suggest that the gap penalties are not optimal. It is important to take the time to get a basic understanding of how a given program works, or it is unlikely to do the best job.
3.3. Algorithm Failure
Clustered alignment is a heuristic strategy that is not guaranteed to find the optimal multiple alignment. The alignment process is likely to compound errors introduced by the user or in the input sequences. Alignment mistakes will also be made in difficult alignment cases, even when there are no input errors. There are many instances of homologous proteins having diverged over long periods of time, so that they have apparently very little sequence similarity. The “twilight zone” where sequence similarity merges with sequence dissimilarity is in the range 20% to 25% identity. (Five percent identity would be a random match for protein sequence, neglecting residue biases. In practice, given residue biases and gap insertions to maximize the similarity, the expectation for random sequence matches approaches ~15% identity, but higher for short or biased sequences. There is, however, no a priori reason why correctly aligned but extremely divergent proteins should not be found with 0% pairwise identity.) Divergent nucleotide sequences are even harder to align, because random similarity is reached at ~25% identity before gaps are added. Wherever possible, alignments need to be checked against additional data available for a sequence family, such as known tertiary structures and whether invariant catalytic residues, or any other known conserved motifs, are correctly aligned.
3.4. Consequences of Errors in Aligned Sequences
Errors in multiple alignments can have disastrous consequences for phylogenetic inference on the basis of sequence trees. Sequences with misaligned segments or translation frameshifts are apparently more divergent than they should be from the other sequences. The branch leading to that sequence will then have a longer length (which erroneously implies a more rapid molecular clock) and the branch point may migrate toward the centre of the tree, giving a false order of divergence.
Such incorrect phylogenies may be quite exciting when they seem to refute established viewpoints. Two rules of thumb are useful: (i) Infinitely more wrong tree topologies can be generated than right ones, and (ii) wrong phylogenies are more interesting than right phylogenies. Incautious advocacy of a wrong phylogeny can waste many peoples' time.
Errors also disrupt evaluation of conserved sites in sequences (Fig. 2). Catalytic residues are often absolutely conserved, so a single misaligned sequence may lead to rejection of the correct site. Most conserved residues have a structural role: Structure prediction for protein uses conserved hydrophobic residues and, for RNA, conserved base-pairing residues. Misalignments disrupt the conservation periodicities, leading to rejection of the correct structures. Terminally truncated sequences of multidomain proteins can lead to false inferences for the domain boundaries, which are very usefully defined by coincidence with the protein N- or C-termini.
References
1. S. B. Needleman and C. D. Wunsch (1970) J. Mol. Biol. 48, 443–453.
2. T. F. Smith and M. S. Waterman (1981) Adv. Appl. Math. 2, 482–489
3. M. O. Dayhoff, R. M. Schwartz, and B. C. Orcutt (1978) In Atlas of Protein Sequence and Structure, Vol. 5, Suppl. 3 (M. O. Dayhoff, ed.), NBRF, Washington, pp. 345–352.
4. S. A. Benner, M. A. Cohen, and G. H. Gonnet (1994) Protein Eng. 7, 1323–1332.
5. G. Vogt, T. Etzold, and P. Argos (1995) J. Mol. Biol. 249, 816–831.
6. O. Gotoh (1982) J. Mol. Biol. 162, 705–708.
7. E. W. Myers and W. Miller (1988) CABIOS 4, 11–17.
8. J. D. Thompson (1995) CABIOS 11, 181–186.
9. J. A. Grice, R. Hughey, and D. Speck (1997) CABIOS, 13, 45–53.
10. M. S. Waterman and M. Eggert (1987) J. Mol. Biol. 197, 723–728.
11. S. F. Altschul, W. Gish, W. Miller, E. W. Myers, and D. J. Lipman (1990) J. Mol. Biol. 215. 403–410 ,
12. W. R. Pearson and D. J. Lipman (1988) Proc. Natl. Acad. Sci. USA 85, 2444–2448.
13. B. Rost and C. Sander (1993) J. Mol. Biol. 232, 584–599.
14. A. Lupas, M. Van Dyke, and J. Stock (1991) Science, 252, 1162–1164.
15. B. Persson and P. Argos (1994) J. Mol. Biol. 237, 182–192.
16. M. Gribskov, A. D. McLachlan, and D. Eisenberg (1987) Proc. Natl. Acad. Sci. USA, 84, 4355-4358 .
17. S. Eddy (1996) Curr. Opin. Struct. Biol. 6, 361–365.
18. P. Bork and T. J. Gibson (1996) Methods Enzymol. 266, 162–184.
19. C. R. Woese, R. R. Gutell, R. Gupta, and H. F. Noller (1983) Microbiol. Rev. 47, 621–669.
20. F. Michel and E. Westhof (1990) J. Mol. Biol. 216, 585–610.
21. R. F. Doolittle (1990) Molecular Evolution: Computer Analysis of Protein and Nucleic Acid Sequences, Methods in Enzymology, Vol. 183, Academic Press, San Diego, CA.
22. D. J. Lipman, S. F. Altschul, and J. D. Kececioglu (1989) Proc. Natl. Acad. Sci USA 86, 44124415- .
23. O. Gotoh (1996) J. Mol. Biol. 264, 823–838.
24. C. Notredame and D. G. Higgins (1996) Nucleic Acids Res. 24, 1515–1524.
25. J. D. Thompson, D. G. Higgins, and T. J. Gibson (1994) Nucleic Acids Res. 22, 4673–4680.
26. D.-F. Feng and R. F. Doolittle (1987) J. Mol. Evol. 25, 351–360.
27. N. Saitou and M. Nei (1987) Mol. Biol. Evol. 4, 406–425.
28. A. M. Lesk, M. Levitt, and C. Chothia (1986) Protein Eng. 1, 77–78.
29. A. Krogh, M. Brown, S. Mian, K. Sjölander, and D. Haussler (1994) J. Mol. Biol. 235, 15011531- .
30. E. Birney, J. D. Thompson, and T. J. Gibson (1996) Nucleic Acids Res. 24, 2730–2739.
31. J. M. Claverie (1993) J. Mol. Biol. 234, 1140–1157.

|
|
|
|
كيف تعزز نمو الشعر الصحي؟
|
|
|
|
|
|
|
10 فحوصات مهمة يجب القيام بها لسيارتك قبل الصيف
|
|
|
|
|
|
قسم الشؤون الفكرية يختتم برنامجاً ثقافياً لوفدٍ من جامعة البصرة
|
|
|
|
جامعة الكفيل تعقد ورشة عمل عن إجراءات عمل اللجان الامتحانيّة
|
|
|
|
قسم التطوير يُقيم دورة أخلاقيّات المهنة ضمن برنامج تأهيل المنتسبين الجدد
|
|
|
|
قسم الشؤون الفكريّة يعلن عن إقامة دورة خاصة بالفتيان خلال العطلة الصيفية
|















