
آخر المواضيع المضافة

 النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية
النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية| Protein Structure |


|
|
Read More
Date: 19-10-2015
Date: 28-10-2015
Date: 14-10-2015
|
Protein Structure
Proteins are chains of amino acids that fold into a three-dimensional shape. Proteins come in a wide variety of amino acid sequences, sizes, and three-dimensional structures, which reflect their diverse roles in nearly all cellular functions. Each protein has a particular structure necessary to bind with a high degree of specificity to one or a few molecules and to carry out its function; thus, function is directly correlated to structure of the protein. Proteins make up about 50 percent of the dry weight of cells and are the most abundant of the macromolecules inside the cell and of the cellular membranes. Proteins (including their lipoprotein and glycoprotein forms) also constitute 10 percent of the weight of the blood plasma of living organisms, carrying various nutrients throughout the body and acting as signals to coordinate bodily functions between the different organs.
The sizes of proteins vary greatly. The size is described by the molecular weight given in the units of a dalton. One dalton is the molecular mass of one hydrogen atom. The molecular weight of a protein is equal to the addition of the molecular weights of the amino acids constituting the protein. Some proteins are of relatively small molecular size, such as insulin, with a molecular weight of about 5,700 daltons. Others, like titin (a protein found in muscle), are very large. Some proteins consist of a single amino acid sequence (polypeptide chain), while others are multimers of the same or different subunits.
Organization of Protein Structure
There are four levels of protein structure: primary, secondary, tertiary, and quaternary. These levels also reflect their temporal sequence. Proteins are synthesized as a primary sequence and then fold into secondary tertiary and quaternary structures. The figures show these various types of structure, which are described as follows.
Amino acids (see Figure 1) are the building blocks (units) of proteins. Each amino acid has several common features: an amino and a carboxyl chemical group both bonded to the alpha carbon (Ca) and an R group that defines a particular amino acid. Some R groups are hydrophobic and tend to project to, and be buried in, the inside of a protein structure. Four amino acid R groups contain either a positive or negative charge and thus project to the water environment to the exterior of proteins. Other R groups are polar in nature and also tend to project to the outside.
The amino acids of a protein are connected to each other by peptide bonds. During protein synthesis the amino group of the amino acid being added is coupled to the carboxyl group of the prior amino acid, and two hydrogen atoms and one oxygen atom are removed as a water molecule (H2O) and the peptide bond is formed (see Figure 2).
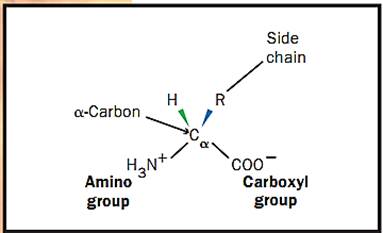
Figure 1. Structure of an amino acid.
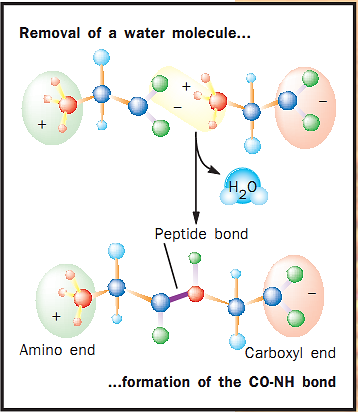
Figure 2. Formation of a peptide bond
Primary Structure of Proteins. The linear sequence of amino acids constitutes a protein’s primary structure. The sequence is written from the amino-terminal end (the first amino acid) to the carboxyl-terminal end (the same sequence in which the protein is synthesized). All properties of a protein are derived from the primary structure, the linear sequence. Encoded in the sequence is the ability of the protein to fold into its secondary, tertiary, and quaternary structures, and thus to be able to carry out a function. The function of a protein is only expressed when the protein has achieved its three-dimensional shape.
Secondary Structures of Proteins. Secondary structures arise from non- covalent interactions between amino acids across the chain. There are only two bonds that can rotate in space between each amino acid in the backbone of the primary sequence. These restricted movements, when repeated through several amino acids in a chain, yield the two main types of protein secondary structure: the alpha (a) helix and the beta 0) strand.
The helix is shaped like a spiral staircase, with each step representing a single amino acid. Each 3.6 amino acids complete a 360-degree turn in the helix. If a helical portion of a protein contained 36 amino acids, there would be 10 complete turns in the helix. Each amino acid projects an R group to the outside of the staircase. A helices of proteins vary in length from 5 to 40 amino acids with an average of about 10. Certain proteins are made up entirely of a helices (and the loops connecting the helices) such as the subunits of hemoglobin, which contain 8 a helices.
As the name “strand” implies, the amino acids of the p strand form a linear structure. However, the bond angles along the peptide backbone produce a regular zigzag pattern within this linear structure. Adjacent R groups project in opposite directions. When amino acid sequences fold into a three-dimensional structure of p strands, one amino acid R group will then project to the interior of the protein and the adjacent R group will project to the outside (to the water environment).
P strands of proteins may be arranged adjacent to each other like strings on an instrument to form what is termed a p sheet. The p strands of a p sheet may be parallel in orientation (all the sequences running from amino- to carboxyl-terminal) or antiparallel (that is, the strands alternate in orientation).
To form a complete protein, the helices or p strands must be joined together through the amino acid sequence. The amino acids that make up these joining regions are called “loops.” For example, two adjacent antiparallel p strands of a p sheet are often connected by a loop consisting of two or three amino acids. Loops also connect segments of a helices and connect p strands that are adjacent to a helices in a protein sequence. Some loop regions can be very long, consisting of up to twenty-one amino acids; but, most commonly, they are between two and ten amino acids.
Tertiary Structures of Proteins. The three-dimensional structure of a single polypeptide chain is termed its tertiary structure. Tertiary structures are different combinations of the secondary structures (a helices, p strands, and loops). Tertiary structure is subdivided into certain portions that are termed motifs and domains.
Motifs are simple combinations of secondary structure that occur in many different proteins and which carry out a similar function. An example is the helix-loop helix. It consists of two antiparallel a helices at about a 60- degree angle to each other connected by a loop. This motif, which binds the calcium ion, is found in several proteins that regulate cellular activity via changes in calcium ion concentrations. Many proteins that bind to deoxyribonucleic acid (DNA) and regulate gene expression incorporate a zinc finger motif. As the name implies, this motif binds the zinc ion using combinations of the amino acids cysteine and histidine. One type of zinc finger motif consists of a single a helix opposite two p strands in an antiparallel arrangement. The zinc ion is held between the helix and the two p strands using two histidine R groups from the helix and two cysteine R groups from the 3 strands. Another motif common to DNA binding proteins is the leucine zipper, in which two parallel a helices are adjacent to each other with the leucine side chains projecting from each helix binding together and thus binding the two helices together.
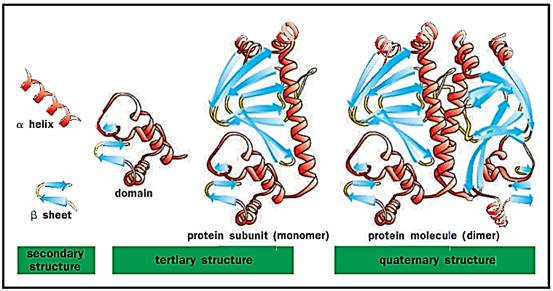
Levels of protein structure. Alpha helices and beta sheets are linked by loop regions to form less-structured domains, which combine to form larger subunits and ultimately functional proteins
A single polypeptide chain may fold into one or more domains to yield the tertiary structure of a protein. The eight a helices of a subunit of hemoglobin connected by seven loop regions constitute the globin domain. Two 3 sheets (each of four antiparallel 3 strands) form a “3 barrel” structure domain that is repeated in the immunoglobulin proteins. Each domain can express a distinct function and is sometimes arranged in a single protein to efficiently carry out an overall function that has several parts. For example, there are seven different chemical reactions that act in sequence to synthesize a fatty acid. In mammals, the fatty acid synthetase enzyme is a single polypeptide chain folded into seven domains, each domain carrying out one of the seven chemical reactions.
Quaternary Structures of Proteins. Two or more polypeptide chains may bind to each other to form a quaternary structure. The quaternary structure of hemoglobin, for example, consists of four polypeptide chains, two α and two β subunits arranged in space in a defined manner.
How Does Protein Structure Determine Function? For almost all biological functions to be expressed, two molecules must bind to each other. An antibody protein must bind to an antigen to provoke an immune response, a hormone protein (for example, a growth factor) must bind to a cell surface receptor to trigger a cell reaction, an enzyme protein must bind to a substrate to catalyze a reaction, and a protein containing the leucine zipper motif must bind to DNA to regulate gene expression. In order for two molecules to bind, they must recognize each other and form a series of noncovalent bonds. Recognition of two molecules for each other is termed “structural complementarity”; that is, the three-dimensional structures must complement each other in the shapes of the interacting surfaces. Analogies that have been used are a key fitting into a lock or the wooden square of a simple child’s game that fits into the square-shaped cutout of a puzzle board.
“Ligand” is the general term used to denote the molecule bound by the protein. When a protein binds to a ligand, many noncovalent bonds are formed. These may be ionic bonds between the charged acidic or basic groups of side chains of amino acids, or hydrogen bonds whereby a hydrogen proton is shared between two atoms or a weak force of binding termed van der Waals bonding that can occur between any two atoms that are very close in space. Water may also be excluded from the surfaces of two molecules binding to each other, contributing what is called the hydrophobic effect. The number of such noncovalent bonds formed between two molecules directly relates to the strength (the affinity) of binding. Thus strength of binding can be strong, as in the case of a protein hormone binding to a cell surface receptor, or weak, as with binding to their substrate enzymes.
Binding of ligands occurs on certain portions of the protein surface. All three types of secondary structure (or combinations of secondary structure) can be involved in binding a particular ligand. The immunoglobulin molecule uses a total of six loop structures, three each from the variable domains of the heavy and light chains to bind to an antigen. By a very large number of variations of the spatial relationships of these loop regions and differences in amino acid residues of the loops, immunoglobulins exhibit binding activities to a very large number of antigens that are encountered in the environment.
For instance, DNA-binding proteins often use a helices to recognize and bind to the nucleic acids of DNA sequences. Different sequences of amino acids along the helices allow such gene regulatory proteins to recognize specific nucleic acid sequences of the DNA and thus to alter expression of a single or only a few genes. The hexokinase protein binds both glucose and ATP to form glucose-phosphate, the first step in the metabolism of glucose through the glycolytic pathway. The hexokinase protein has two domains, and the glucose spatially complements within a groove between the two domains and thus is bound by the enzyme. Galactose is another sugar very similar to glucose except for the spatial orientation of one of five hydroxyl groups common to glucose and galactose. This single hydroxyl orientation difference does not allow galactose to bind to hexokinase and thus hexokinase exhibits specificity of binding. Galactose is phospho- rylated by another enzyme protein, galactokinase, which exhibits specificity for the galactose sugar; that is, galactokinase structurally complements and binds galactose, but not glucose.
Protein Modifications
Proteins can be glycosylated (glycoproteins) or associated with lipids (lipoproteins).
Glycoproteins. Glycoproteins have attached carbohydrate molecules (residues). Carbohydrate residues are added to the protein structure and modified during and following protein synthesis. There are many different carbohydrate sequences found in glycoproteins, many of which have functional consequences. In general, most proteins that are secreted from cells are glycosylated. Most of the proteins in serum are glycosylated as are the proteins found in saliva and the digestive juices of the gastrointestinal tract. Carbohydrates have many hydroxyl (-OH) groups that bind to water molecules, and thus increase stability. Thus the glycoproteins of saliva tend to lubricate the food chewed, in part to allow easier swallowing of food and its passage through the esophagus. The glycoproteins secreted in the stomach protect the lining of that organ from its acidic environment. This protective role of carbohydrates is also apparent for the serum glycoproteins. The carbohydrates on the surface of the protein protect the protein from the actions of proteases that degrade protein structures.
Certain types of carbohydrate residues on glycoproteins also serve as signal mechanisms. If a tissue is injured or becomes infected, certain glycoproteins are recruited to the surfaces of endothelial cells (the cells that line all blood vessels), where they are recognized by white blood cells, as a signal that this is a site of injury requiring attention. Particularly in the last ten years, about three hundred functions of the carbohydrate portions of glycoproteins have been described.
Lipoproteins. Lipoproteins are complexes between a particular set of proteins (termed apoproteins) and lipids (phospholipids, triglycerides, cholesterol, and cholesterol esters). These lipids are transported throughout the body as the complex lipoproteins. Humans have nine different apoproteins of various molecular sizes and concentrations in the blood. Portions of the surfaces of apoproteins exhibit specificity and bind the various lipids, providing the scaffold upon which the lipoprotein particles are constructed. Their densities—high-density (HDL), low-density (LDL), and very-low- density lipoproteins (VLDL)—are used to characterize the protein. The apoproteins are synthesized in the cell (mainly in the liver) and acquire lipids to become HDL, LDL, or VLDL.
Proteins and Evolution
The presence of similar domain structures in different proteins, the duplication of domain structures in a single protein, and similarities in amino acid sequences (sequence homologies) indicate an evolutionary relationship of many proteins in a single species and between species. There are many examples of these relationships, of which a few will be described here.
The globin fold, as described above, consists of eight a helices (connected by loops) that form a pocket as an active site. A heme structure is bound in many globin fold proteins that binds and carries oxygen in an organism. The globin fold structure has been preserved in mammals, insects, and plants although the amino acid sequence similarities may be very low between such disparate species. Thus, natural selection has maintained similar structures to carry out similar functions even as the gene sequences have diverged to such a great extent between the different species.
The helix-turn-helix motif is common to many gene repressor proteins that bind to DNA sequences. Rigorous statistical analyses of the amino acid sequences of these motifs suggest that these repressor proteins all evolved from a common ancestral gene and that certain amino acid residues in the motif structure are crucial to maintain the helix-turn-helix structure of the motif.
Serine proteases (for example, chymotrypsin, a digestive enzyme in mammals) consist of two p barrel domains, the ends of which come together to form an active site. Within the active site is a catalytic triad, which consists of three amino acids (histidine, serine, and cysteine) arranged in space to catalyze the hydrolysis of a peptide bond. The two p barrels probably evolved from duplication of a common gene. In humans, there are many serine proteases that cleave peptide bonds of different proteins. All have the same two p barrel domain structure with the same spatial catalytic triad. Specificity of binding and cleaving different proteins is achieved by altering the sequences around the catalytic triad such that different proteins complement the different binding sites.
Protein Structures and Disease
Some differences of amino acid sequences of proteins are directly related to disease. A well-defined example is that of sickle-cell disease. A single difference at position number 6 in the amino acid sequence of the p chain of hemoglobin (a valine amino acid is found in the person with sickle-cell disease instead of glutamic acid) results in aggregation of the hemoglobin molecules with consequent elongation (the sickle shape) and fragility of the red blood cells. The disease cystic fibrosis has now been defined as mutations in a particular gene that codes for a cell membrane protein that functions to pump chloride ions out of the cell. This protein in cystic fibrosis is defective in this function because the amino acid sequence is different from normal.
References
Alberts, Bruce, et al. Molecular Biology of the Cell, 4th ed. New York: Garland Publishing, 2000.
Branden, Carl, and John Tooze. Introduction to Protein Structure. New York and London: Garland Publishing, Inc., 1997.
Stryer, Lubert. Biochemistry, 4th ed. New York: W. H. Freeman and Company, 1995.

|
|
|
|
"عادة ليلية" قد تكون المفتاح للوقاية من الخرف
|
|
|
|
|
|
|
ممتص الصدمات: طريقة عمله وأهميته وأبرز علامات تلفه
|
|
|
|
|
|
|
المجمع العلمي للقرآن الكريم يقيم جلسة حوارية لطلبة جامعة الكوفة
|
|
|
















