آخر المواضيع المضافة

 علم الكيمياء
الكيمياء التحليلية
الكيمياء الحياتية
الكيمياء العضوية
الكيمياء الفيزيائية
الكيمياء اللاعضوية
مواضيع اخرى في الكيمياء
الكيمياء الصناعية
علم الكيمياء
الكيمياء التحليلية
الكيمياء الحياتية
الكيمياء العضوية
الكيمياء الفيزيائية
الكيمياء اللاعضوية
مواضيع اخرى في الكيمياء
الكيمياء الصناعية | The Nonclassical Carbocation Hypothesis |


|
|
Read More
Date: 3-8-2019
Date: 21-7-2019
Date: 11-9-2018
|
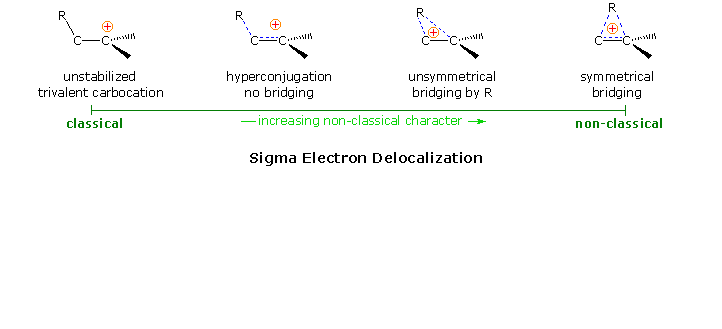
The role of carbocation intermediates in many organic reactions is well established. Some, such as tert-butyl, are localized. Some,such as allyl and benzyl, are stabilized by conjugation to pi-electron systems. Some, as described above, are stabilized by bridging to neighboring nucleophiles. In all cases of anchimeric assistance described above, a charge delocalized or redistributed species is an intermediate on the reaction path. Such intermediates can be isolated in some cases, but they usually have only transitory existence. The rate acceleration of ionization is attributed to structural and energetic similarities of the transition states to the intermediates they produce (the Hammond postulate).
Anchimeric assistance is usually associated with one or more of the following observable characteristics.
• Rate acceleration compared with similar reactions lacking assistance.
• Stereoelectronic control that results in rate and product differences between stereoisomers.
• Retention of configuration in substitution products.
• Racemization of products (and often reactants) when a symmetrical bridged intermediate is involved.
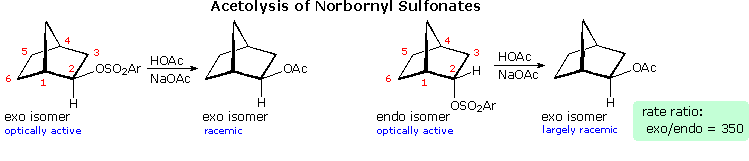
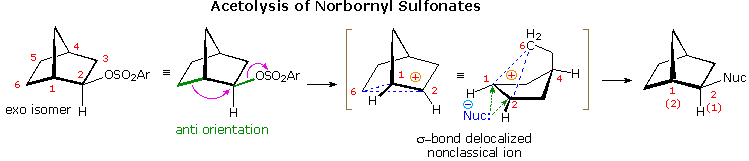
Solvolysis of the exo and endo-2-norbornyl sulfonate esters disclosed differences that suggested anchimeric assistance for the exo-isomer. As shown in the following diagram, the rate of acetolysis of the exo-isomer is substantially faster than that of the endo-isomer, which reacts at a rate similar to the cyclohexyl derivative. The former substitution proceeds with complete retention of configuration and racemization; whereas the endo-isomer is substituted with inversion of configuration and retains a small degree of optical activity. The source of this assistance was proposed to be the electron pair of the C1 : C6 sigma bond, which is ideally oriented anti to the sulfonate leaving group.
Since this bridged ion is symmetrical, formation of racemic acetate is expected. The term "nonclassical" was applied to this charge delocalized cation, inasmuch as it appeared to be unique.
By comparison, the endo-isomer ionizes to a classical 2º-carbocation, which is rapidly converted to the more stable nonclassical ion. Some acetate anion may bond to the 2º-carbocation before it changes, accounting for the residual optical activity in this reaction.
Not everyone was convinced by this interpretation of the evidence. The chief protagonists favoring the nonclassical view were S. Winstein and J. D. Roberts. The primary opposition came from H. C. Brown, who espoused a more conventional rationalization. Brown pointed out that the norbornyl compounds are better compared with cyclopentyl than with cyclohexyl analogs (eclipsing strain), and in such a comparison the endo isomer is abnormally slow, the exo isomer being only 14 times faster than cyclopentyl. The racemic product was explained by assuming the interconversion of enantiomeric classical carbocations was very rapid on the reaction time scale. Brown also noted that attachment of a stabilizing aryl substituent at C2 did not reduce the rate enhancement of exo-ionization or the preference for exo-product formation. Since these latter solvolyses proceed by way of a benzylic cation, sigma-bond assistance was assumed to be minimal. Consequently, rate enhancement and retention of configuration become less significant as nonclassical indicators. This latter experiment, in which the aryl substituent was p-anisyl (An), is depicted on the left side of the diagram below.
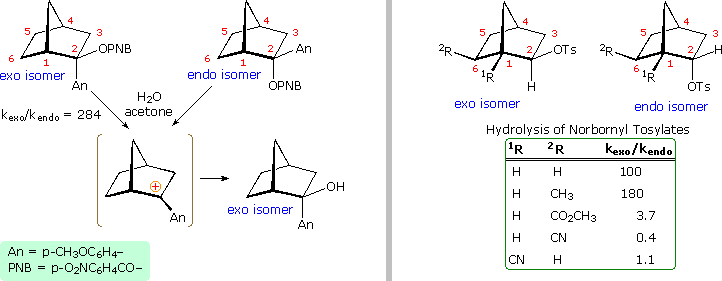
Despite Brown's damaging arguments, other experiments provided additional support for the nonclassical view. As shown on the right side of the diagram, electron withdrawing substituents on C6 (2R) retarded exo-reactivity more severely than endo-reactivity. A similar effect was noted for such substituents at C1 (1R). This influence is best explained by the nonclassical hypothesis, in which partial positive charge must be carried by C1, C2 & C6.
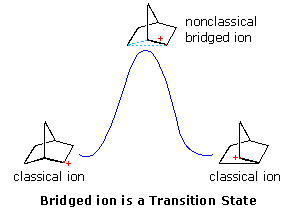
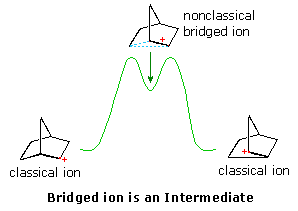
Interpretations of the considerable body of evidence amassed at this point may be summarized in the diagram on the right. In the first display, the nonclassical bridged cation is shown as a transition state for the interconversion of the classical carbocations. A relationship of this kind corresponds to the rearrangement of neopentyl chloride. A second possibility, presented by clicking on the diagram, has the nonclassical ion as a higher energy intermediate, linking the classical ions. Finally, by clicking on the diagram a second time, the possibility that the nonclassical ion represents the more stable intermediate is drawn.
By the mid 1960's chemical and nmr techniques had improved to a stage that allowed direct observation of carbocations in low nucleophilic, acidic solutions, often referred to as "super acids". Much of this work was conducted by George Olah (Nobel Prize, 1994), using mixed solvents composed of SbF5, SO2, SO2F2 & SO2FCl. At low temperatures, 1H and 13C nmr spectra of (CH3)3C(+) and (CH3)2CH(+) were obtained and interpreted. As anticipated, the charged tricoordinate carbon atom exhibited a 13C signal over 300ppm downfield from TMS. When similar nmr measurements were applied to the 2-norbornyl cation, a number of fast proton shifts were disclosed. These could be "frozen out" by working at low temperature, the 3,2-shift at -70º C and a faster 6,2-shift at -130º C. The resulting spectrum, which remained unchanged at temperatures as low as -160º C, had no low field signals near that expected for a classical 2º-carbocation, and was supportive of the nonclassical structure. Recently, a solid state 13C nmr spectra at 5º K proved consistent with the nonclassical ion. From these and other spectroscopic studies, the sigma-bridged nonclassical cation has been firmly identified as the more stable carbocation species having the 2-norbornyl structure. Further confirmation was provided in 2013 by researchers in Germany, employing careful X-ray crystallographic measurements of an annealed [C7H11]+[Al2Br7]– salt at 40º K.
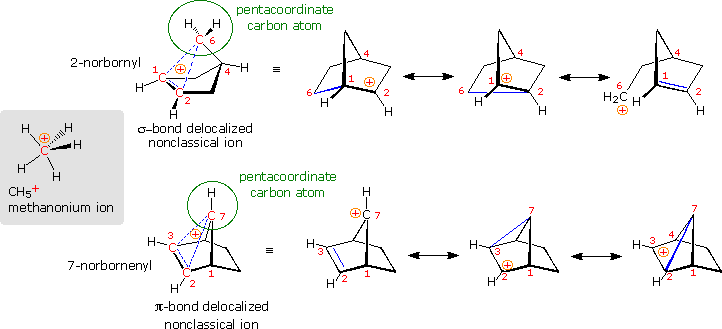
Are there other relatively stable nonclassical carbocations? Several that seem to fit this classification have been identified, but few have been as exhaustively studied as the 2-norbornyl. One of the best criteria for evaluating candidate ions is to establish whether one or more of the participating carbon atoms is hypervalent (has more than four coordinating groups). In the following diagram, the simplest hypervalent carbocation, methanonium, is drawn on the left in the gray shaded box. This ion is commonly seen in the mass spectrum of methane (gas phase), but decomposes in solution as a consequence of its extreme acidity. To its right are two larger non-classical ions, 2-norbornyl and 7-norbornenyl. A pentacoordinate carbon atom is identified in each case. Resonance contributors to these ions are shown to the right of the dashed bond representation, and in all the drawings the delocalized electron pair is colored blue.


|
|
|
|
"علاج بالدماغ" قد يخلص مرضى الشلل من الكرسي المتحرك
|
|
|
|
|
|
|
تقنية يابانية مبتكرة لإنتاج الهيدروجين
|
|
|
|
|
|
|
المجمع العلمي يطلق مشروع (حفظة الذكر) في قضاء الهندية
|
|
|