























النبات

مواضيع عامة في علم النبات
الجذور - السيقان - الأوراق
النباتات الوعائية واللاوعائية
البذور (مغطاة البذور - عاريات البذور)
الطحالب
النباتات الطبية
الحيوان
مواضيع عامة في علم الحيوان
علم التشريح
التنوع الإحيائي
البايلوجيا الخلوية
الأحياء المجهرية
البكتيريا
الفطريات
الطفيليات
الفايروسات
علم الأمراض
الاورام
الامراض الوراثية
الامراض المناعية
الامراض المدارية
اضطرابات الدورة الدموية
مواضيع عامة في علم الامراض
الحشرات
التقانة الإحيائية
مواضيع عامة في التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحيوية والميكروبات
الفعاليات الحيوية
وراثة الاحياء المجهرية
تصنيف الاحياء المجهرية
الاحياء المجهرية في الطبيعة
أيض الاجهاد
التقنية الحيوية والبيئة
التقنية الحيوية والطب
التقنية الحيوية والزراعة
التقنية الحيوية والصناعة
التقنية الحيوية والطاقة
البحار والطحالب الصغيرة
عزل البروتين
هندسة الجينات
التقنية الحياتية النانوية
مفاهيم التقنية الحيوية النانوية
التراكيب النانوية والمجاهر المستخدمة في رؤيتها
تصنيع وتخليق المواد النانوية
تطبيقات التقنية النانوية والحيوية النانوية
الرقائق والمتحسسات الحيوية
المصفوفات المجهرية وحاسوب الدنا
اللقاحات
البيئة والتلوث
علم الأجنة
اعضاء التكاثر وتشكل الاعراس
الاخصاب
التشطر
العصيبة وتشكل الجسيدات
تشكل اللواحق الجنينية
تكون المعيدة وظهور الطبقات الجنينية
مقدمة لعلم الاجنة
الأحياء الجزيئي
مواضيع عامة في الاحياء الجزيئي
علم وظائف الأعضاء
الغدد
مواضيع عامة في الغدد
الغدد الصم و هرموناتها
الجسم تحت السريري
الغدة النخامية
الغدة الكظرية
الغدة التناسلية
الغدة الدرقية والجار الدرقية
الغدة البنكرياسية
الغدة الصنوبرية
مواضيع عامة في علم وظائف الاعضاء
الخلية الحيوانية
الجهاز العصبي
أعضاء الحس
الجهاز العضلي
السوائل الجسمية
الجهاز الدوري والليمف
الجهاز التنفسي
الجهاز الهضمي
الجهاز البولي
المضادات الميكروبية
مواضيع عامة في المضادات الميكروبية
مضادات البكتيريا
مضادات الفطريات
مضادات الطفيليات
مضادات الفايروسات
علم الخلية
الوراثة
الأحياء العامة
المناعة
التحليلات المرضية
الكيمياء الحيوية
مواضيع متنوعة أخرى
الانزيمات

Glycoprotein Synthesis
 المؤلف:
Denise R. Ferrier
المؤلف:
Denise R. Ferrier
 المصدر:
Lippincott Illustrated Reviews: Biochemistry
المصدر:
Lippincott Illustrated Reviews: Biochemistry
 الجزء والصفحة:
الجزء والصفحة:
 3-10-2021
3-10-2021
 2946
2946




+
-
20
Glycoprotein Synthesis
Proteins destined to function in the cytoplasm are synthesized on free cytosolic ribosomes. However, proteins, including glycoproteins, that are destined for cellular membranes, lysosomes, or to be exported from the cell, are synthesized on ribosomes attached to the RER. These proteins contain specific signal sequences that act as molecular addresses, targeting the proteins to their proper destinations. An N-terminal hydrophobic sequence initially directs these proteins to the RER, allowing the growing polypeptide to be extruded into the lumen .
The proteins are then transported via secretory vesicles to the Golgi, which acts as a sorting center (Fig. 1). In the Golgi, those glycoproteins that are to be secreted from the cell (or are targeted for lysosomes) are packaged into vesicles that fuse with the cell (or lysosomal) membrane and release their contents. Those that are destined to become components of the cell membrane are integrated into the Golgi membrane, which buds off, forming vesicles that add their membrane-bound glycoproteins to the cell membrane. [Note: Therefore, the membrane glycoproteins are oriented with the carbohydrate portion on the outside of the cell (see Fig. 1).]

Figure 14.15 Transport of glycoproteins to and through the Golgi and their subsequent secretion or incorporation into a lysosome or the cell membrane.
A. Carbohydrate components
The precursors of the carbohydrate components of glycoproteins are nucleotide sugars, which include UDP-glucose, UDP-galactose, UDPGlcNAc, and UDP-GalNAc. In addition, guanosine diphosphate (GDP)-mannose, GDP-L-fucose (which is synthesized from GDP-mannose), and CMP-NANA may donate sugars to the growing chain. [Note: When the acidic NANA is present, the oligosaccharide has a negative charge at physiologic pH.] The oligosaccharides are covalently attached to the side chains of specific amino acids in the protein, where the three-dimensional structure of the protein determines whether or not a specific amino acid is glycosylated.
B. O-Linked glycoprotein synthesis
Synthesis of the O-linked glycoproteins is very similar to that of the GAG . First, the protein to which sugars are to be attached is synthesized on the RER and extruded into its lumen. Glycosylation begins with the transfer of GalNAc (from UDP-GalNAc) to the hydroxyl group of a specific serine or threonine residues. The glycosyltransferases responsible for the stepwise synthesis (from individual sugars) of the oligosaccharides are bound to the membranes of the Golgi. They act in a specific order, without using a template as is required for DNA, ribonucleic acid (RNA), and protein synthesis (see Unit VII), but instead by recognizing the actual structure of the growing oligosaccharide as the appropriate substrate.
C. N-Linked glycoprotein synthesis
Synthesis of N-linked glycoproteins occurs in the lumen of the RER and requires the participation of the phosphorylated form of dolichol (dolichol pyrophosphate), a lipid of the RER membrane (Fig. 2). The initial product is processed in the RER and Golgi.
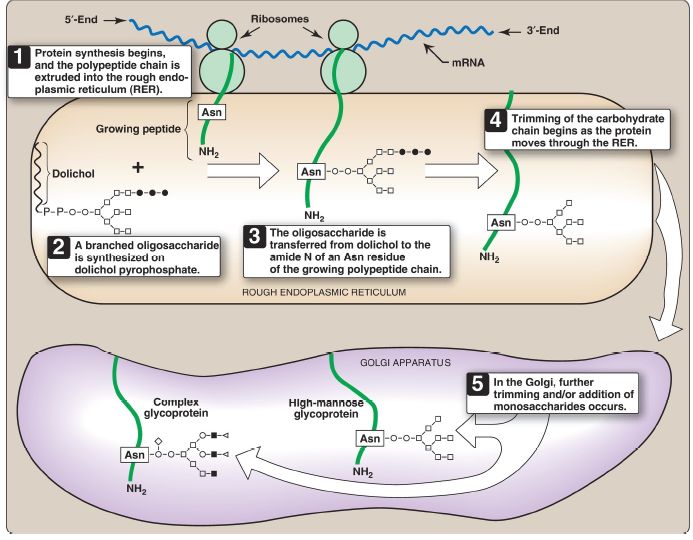
Figure 2: Synthesis of N-linked glycoproteins. ○ = N-acetylglucosamine; = mannose; • = glucose; ▪ = galactose; ◇ or ► = terminal group (fucose or Nacetylneuraminic acid); mRNA = messenger RNA; Asn = asparagine.
1. Dolichol-linked oligosaccharide synthesis: As with the O-linked glycoproteins, the protein is synthesized on the RER and enters its lumen. However, it does not become glycosylated with individual sugars.
Instead, a lipid-linked oligosaccharide is first constructed. This consists of dolichol (an RER membrane lipid made from an intermediate of cholesterol synthesis) attached through a pyrophosphate linkage to an oligosaccharide containing GlcNAc, mannose, and glucose.
The sugars to be added sequentially to the dolichol by membrane-bound glycosyltransferases are first GlcNAc, followed by mannose and glucose (see Fig. 2). The entire 14-sugar oligosaccharide is then transferred from dolichol to the amide nitrogen of an asparagine residue in the protein to be glycosylated by a protein–oligosaccharide transferase present in the RER. [Note: Tunicamycin inhibits N-linked glycosylation.]
Congenital disorders of glycosylation (CDG) are syndromes caused primarily by defects in the N-linked glycosylation of proteins, either oligosaccharide assembly (type I) or processing (type II).
2. N-Linked oligosaccharide processing: After addition to the protein, the N-linked oligosaccharide is processed by the removal of specific mannosyl and glucosyl residues as the glycoprotein moves through the RER. Finally, the oligosaccharide chains are completed in the Golgi by addition of a variety of sugars (for example, GlcNAc, GalNAc, and additional mannoses and then fucose or NANA as terminal groups) to produce a complex glycoprotein. Alternatively, they are not processed further, leaving branched, mannose-containing chains in a high-mannose glycoprotein (see Fig. 2). The ultimate fate of N-linked glycoproteins is the same as that of the O-linked glycoproteins (for example, they can be released by the cell or become part of a cell membrane). In addition, N-linked glycoproteins can be targeted to the lysosomes. [Note: Nonenzymatic glycosylation of proteins is known as glycation .]
3. Lysosomal enzymes: N-Linked glycoproteins being processed in the Golgi can be phosphorylated on carbon 6 of one or more mannosyl residues. UDP-GlcNAc provides the phosphate in a reaction catalyzed by a phosphotransferase. Receptors, located in the Golgi membrane, bind the mannose 6-phosphate residues of these proteins, which are then packaged into vesicles and sent to the lysosomes (Fig. 3). I-Cell disease is a rare lysosomal storage disease in which the phosphotransferase is deficient. This causes the proteins to be secreted, rather than being targeted to lysosomes. Consequently, the acid hydrolases normally found in the lysosomal matrix are absent, resulting in an accumulation of the substrates for these missing enzymes. [Note: ICell disease is so named because of the large inclusion bodies seen in cells of patients with this disease.] In addition, high amounts of lysosomal enzymes are found in the patient’s plasma and urine, indicating that the targeting process to lysosomes (rather than the synthetic pathway of these enzymes) is deficient. I-Cell disease is characterized by skeletal abnormalities, restricted joint movement, coarse (dysmorphic) facial features, and severe psychomotor impairment. [Note: Because I-cell disease has features in common with the mucopolysaccharidoses and sphingolipidoses , it is termed a mucolipidosis (type II).] Currently, there is no cure, and death from cardiopulmonary complications usually occurs in early childhood.
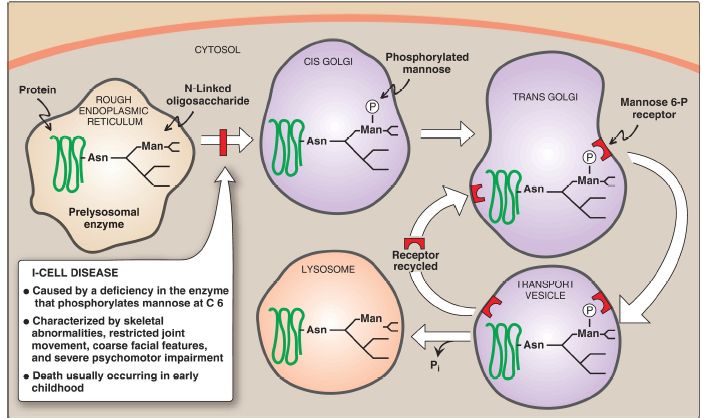
Figure 3: Mechanism for transport of N-linked glycoproteins to the lysosomes. Asn = asparagine; Man = mannose; = phosphate; Pi = inorganic phosphate.
 الاكثر قراءة في الكيمياء الحيوية
الاكثر قراءة في الكيمياء الحيوية
 اخر الاخبار
اخر الاخبار
اخبار العتبة العباسية المقدسة

الآخبار الصحية


مواضيع ذات صلة
 قسم الشؤون الفكرية يصدر كتاباً يوثق تاريخ السدانة في العتبة العباسية المقدسة
قسم الشؤون الفكرية يصدر كتاباً يوثق تاريخ السدانة في العتبة العباسية المقدسة "المهمة".. إصدار قصصي يوثّق القصص الفائزة في مسابقة فتوى الدفاع المقدسة للقصة القصيرة
"المهمة".. إصدار قصصي يوثّق القصص الفائزة في مسابقة فتوى الدفاع المقدسة للقصة القصيرة (نوافذ).. إصدار أدبي يوثق القصص الفائزة في مسابقة الإمام العسكري (عليه السلام)
(نوافذ).. إصدار أدبي يوثق القصص الفائزة في مسابقة الإمام العسكري (عليه السلام)



















