DNA Separation Techniques
KEY CONCEPTS
-Gel electrophoresis separates DNA fragments by size, using an electric current to cause the DNA to migrate toward a positive charge.
-DNA can also be isolated using density gradient centrifugation.
With a few exceptions, the individual pieces of DNA (chromosomes) making up a living organism’s genome are on the order of Mb in length, making them too physically large to be manipulated easily in the laboratory. Individual genes or chromosomal regions of interest by contrast are often quite small and readily manageable, on the order of hundreds or a few thousand bp in length. A necessary first step, therefore, in many experimental processes investigating a specific gene or region, is to break the large original chromosomal DNA molecule down into smaller manageable pieces and then begin isolation and selection of the particular relevant fragment or fragments of interest. This breakage can be done by mechanical
shearing of chromosomes, in a process that produces breakages randomly to produce a uniform size distribution of assorted molecules. This approach is useful if randomness in breakpoints is
required, such as to create a library of short DNA molecules that “tile” or partially overlap one another while together representing a much larger genomic region, such as an entire chromosome or genome. Alternatively, restriction endonucleases can be employed to cut large
DNA molecules into defined shorter segments in a way that is reproducible. This reproducibility is frequently useful, in that a DNA section of interest can be identified in part by its size. Consider a hypothetical gene, genX, on a bacterial chromosome, with the entire gene lying between two EcoRI sites spaced 2.3 kb apart. Digestion of the bacterial DNA with EcoRI will yield a range of small DNA molecules, but genX will always occur on the same 2.3-kb fragment. Depending on the size and complexity of the starting genome, there might be several other DNA segments of similar size produced, or in a simple enough system, this 2.3-kb size might be unique to the genX fragment. In this latter case, detection or visualization of a 2.3-kb fragment is enough to definitively identify the presence of genX. Many of the earliest laboratory techniques developed in working with DNA relate to separating and concentrating DNA molecules based on size expressly to take advantage of these concepts. The ability to separate DNA molecules based on size allows for taking a complex mixture of many fragment sizes and selecting a much smaller, less complex subset of interest for further study.
The simplest method for separation and visualization of DNA molecules based on size is gel electrophoresis. In neutral agarose gel (the most basic type of gel), electrophoresis is done by preparing a small slab of gel in an electrically conductive, mildly basic buffer. Although similar to the gelatins used to make dessert dishes, this type of gel is made from agarose, a polysaccharide that is derived from seaweed and has very uniform molecular sizes.
Preparation of agarose gels of a specific percentage of agarose by mass (usually in the range of 0.8%–3%) creates, in effect, a molecular sieve, with a “mesh” pore size being determined by the percentage of agarose (higher percentages yielding smaller pores).
The gel is poured in a molten state into a rectangular container, with discrete wells being formed near one end of the product. After cooling and solidifying, the slab is submerged in the same conductive, mildly alkaline buffer and samples of mixed DNA fragments are placed in the preformed wells. A DC electric current is then applied to the gel, with the positive charge being at the opposite end of the gel from the wells. The alkalinity of the solution ensures that the DNA molecules have a uniform negative charge from their backbone phosphates, and the DNA fragments begin to be drawn electrostatically toward the positive electrode. Shorter DNA fragments are able to move through the agarose pores with less resistance than longer fragments, and so over time the smallest DNA molecules move the farthest from the wells and the largest move the least. All fragments of a given size will move at about the same rate, effectively concentrating any population of equal-sized molecules into a discrete band at the same distance from the well. The addition of a DNA-binding fluorescent dye to the gel, such as ethidium bromide or SYBR green, stains these DNA bands such that they can be directly seen by eye when the gel is exposed to fluorescence-exciting light. In practice, a standard sample consisting of a set of DNA molecules of a known size is run in one of the wells, with sizes of bands in other wells estimated in comparison to the standard, as shown in FIGURE 1. DNA
molecules of roughly 50 to 10,000 bp can be quickly separated, identified, and sized to within about 10% accuracy by this simple method, which remains a common laboratory technique. DNA molecules can be separated not only by size but also by shape.
Supercoiled DNA, which is compact compared to relaxed or linear DNA, migrates more rapidly on a gel, and the more supercoiling, the faster the migration, as shown in FIGURE 2.
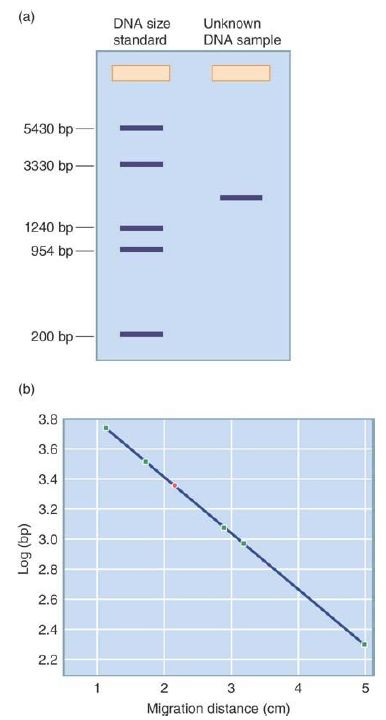
FIGURE 1. DNA sizes can be determined by gel electrophoresis. (a) A DNA of standard size and a DNA of unknown size are run in two lanes of a gel, depicted schematically. (b) The migration of the DNAs of known size in the standard is graphed to create a standard curve (migration distance in cm versus log bp). The point shown in green is for the DNA of unknown size.
Data from an illustration by Michael Blaber, Florida State University.

FIGURE 2. Supercoiled DNA molecules separated by agarose gel electrophoresis. Lane 1 contains untreated negatively supercoiled DNA (lower band). Lanes 2 and 3 contain the same DNA that was treated with a type 1 topoisomerase for 5 and 30 minutes, respectively. The topoisomerase makes a single-strand break in the DNA and relaxes negative supercoils in single steps (one supercoil relaxed per strand broken and reformed).
Reproduced from: Keller, W. 1975. Proc Natl Acad Sci USA 72:2550–2554. Photo courtesy
of Walter Keller, University of Basel.
Variations on this method primarily relate to changing the gel matrix from agarose to other molecules such as synthetic polyacrylamides, which can have even more precisely controlled pore sizes. These can offer finer size resolution of DNA molecules from roughly 10 to 1,500 base pairs in size. Both resolution and sensitivity are further improved by making these types of gels as thin as possible, normally requiring that they be formed between glass plates for mechanical strength. When chemical denaturants such as urea are added to the buffer system, the DNA molecules are forced to unfold (losing any secondary structures) and take on hydrodynamic properties related only to molecule length. This approach can clearly resolve DNA molecules differing in length by only a single nucleotide. Denaturing polyacrylamide electrophoresis is a key component of the classic DNA sequencing technique whereby the separation and detection of a series of single nucleotide–length difference DNA products allows for the reading of the underlying order of nucleotide bases. Another method for separating DNA molecules from other contaminating biomolecules, or in some cases for fractionation of specific small DNA molecules from other DNAs, is through the use of gradients, as depicted in FIGURE3. The most frequent implementation of this is isopycnic banding, which is based on the fact that specific DNA molecules have unique densities based on their G-C content. Under the influence of extreme g-forces, such as through ultracentrifugation, a high-concentration solution of a salt (such as cesium chloride) will form a stable density gradient from low density (near top of tube/center of rotor) to high density (near
bottom of tube/outside of rotor). When placed on top of this gradient (or even mixed uniformly within the gradient) and subjected to continued centrifugation, individual DNA molecules will migrate to a position in the gradient where their density matches that of the surrounding medium. Individual DNA bands can then be either visualized (e.g., through the incorporation of DNA-binding fluorescent dyes in the gradient matrix and exposure to fluorescence excitation) or recovered by careful puncture of the centrifuge tube and fractional collection of the tube contents. This method can also be used to separate double-stranded from singlestranded molecules and RNA from DNA molecules, again based solely on density differences.

FIGURE 3. Gradient centrifugation separates samples based on their density.
Choice of the gradient matrix material, its concentration, and the centrifugation conditions can influence the total density range separated by the process, with very narrow ranges being used to fractionate one particular type of DNA molecule from others, and wider ranges being used to separate DNAs in general from other biomolecules. Historically, one of the best known uses of this technique was in the Meselson–Stahl experiment of 1958 (introduced in the Genes Are DNA and Encode RNAs and Polypeptides chapter), in which the stepwise density changes in the DNA genomes of bacteria shifted from growth in “heavy” nitrogen (15 N) to “regular” nitrogen (14 N) were observed. The method’s capacity to differentially band DNA with pure 15N, half 15N/half 14N, and pure 14N conclusively demonstrated the semiconservative nature of DNA replication. Now, the method is most frequently employed as a large-scale preparative purification technique with wider density ranges to purify DNAs as a group away from proteins and RNAs.





















