Prions Cause Diseases in Mammals
KEY CONCEPTS
-The protein responsible for scrapie exists in two forms: the wild-type noninfectious form PrPc , which is susceptible to proteases, and the disease-causing PrPsc , which is resistant to proteases.
- The neurological disease can be transmitted to mice by injecting the purified PrPsc protein into mice.
- The recipient mouse must have a copy of the PrPsc gene coding for the mouse protein.
- The PrPsc protein can perpetuate itself by causing the newly synthesized PrP protein to take up the PrP form instead of the PrPc form.
- Multiple strains of PrPsc may have different conformations of the protein.
Prion diseases have been found in humans, sheep, cows, and, more recently, in wild deer and elk. The basic phenotype is an ataxia—a neurodegenerative disorder that is manifested by an inability to remain upright. The name of the disease in sheep, scrapie, reflects the phenotype: The sheep rub against walls in order to stay upright. Scrapie can be perpetuated by inoculating sheep with tissue extracts from infected animals. In humans, the disease kuru was found in New Guinea, where it appeared to be perpetuated by cannibalism, in particular the eating of brains. Related diseases in Western populations with a pattern of genetic transmission include Gerstmann–Straussler syndrome and the related Creutzfeldt–Jakob disease (CJD), which occurs sporadically. A disease resembling CJD appears to have been transmitted by consumption of meat from cows suffering from “mad cow” disease.
When tissue from scrapie-infected sheep is inoculated into mice, the disease occurs in a period ranging from 75 to 150 days. The active component is a protease-resistant protein. The protein is encoded by a gene that is normally expressed in the brain. The form of the protein in a normal brain, called PrPc , is sensitive to proteases. Its conversion to the resistant form, called PrPsc , is associated with occurrence of the disease. Neurotoxicity is mediated by PrPL , which is catalyzed by PrPsc and occurs when the PrPL concentration becomes too high. Rapid propagation
results in severe neurotoxicity and eventual death. The infectious preparation has no detectable nucleic acid, is sensitive to UV irradiation at wavelengths that damage protein, and has a low infectivity (1 infectious unit/105 PrPsc proteins). This corresponds to an epigenetic inheritance in which there is no change in genetic information (because normal and diseased cells have the same PrP gene sequence), but the PrPsc form of the protein is the infectious agent (whereas PrPc is harmless). The PrPsc form has a high content of β-sheets, which form an amyloid fibrillous structure that is absent from the PrPc form. The basis for the difference between the PrPsc and PrPc forms appears to lie with a change in conformation rather than with any covalent alteration. Both proteins are glycosylated and linked to the membrane by a glycosylphosphatidylinositol (GPI) linkage.
The assay for infectivity in mice allows the dependence on protein sequence to be tested. FIGURE 1 illustrates the results of some critical experiments. In the normal situation, PrPsc protein extracted from an infected mouse will induce disease (and ultimately kill) when it is injected into a recipient mouse. If the PrP gene is deleted, a mouse becomes resistant to infection. This experiment demonstrates two things. First, the endogenous protein is necessary for an infection, presumably because it provides the raw material that is converted into the infectious agent. Second, the cause of disease is not the removal of the PrPc form of the protein, because a mouse with no PrPc survives normally: The disease is caused by a gain of function in PrPsc . If the PrP gene is altered to prevent the GPI linkage from occurring, mice infected with PrP do not develop disease, which suggests that the gain of function involves an altered signaling function for which the GPI linkage is required.
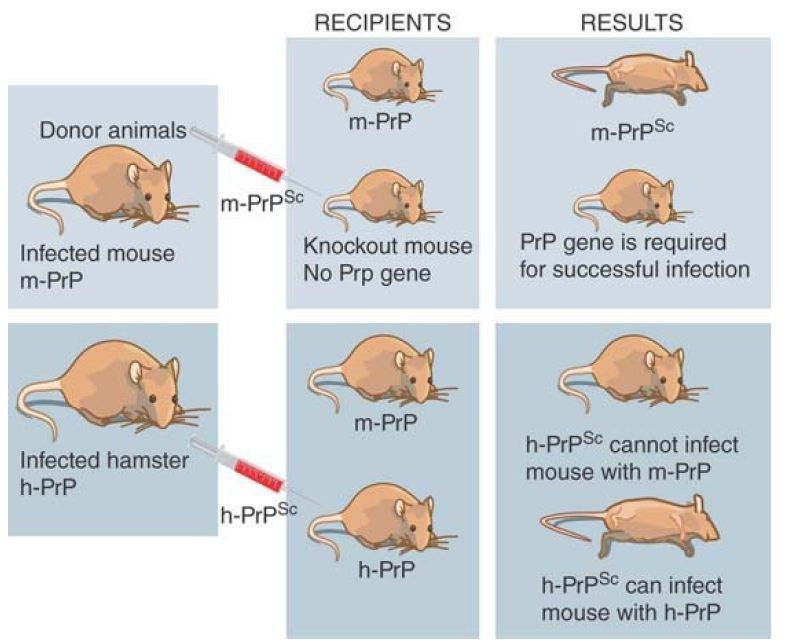
FIGURE 1. A PrPsc protein can only infect an animal that has the same type of endogenous PrPc protein.
The existence of species barriers allows hybrid proteins to be constructed to delineate the features required for infectivity. The original preparations of scrapie were perpetuated in several types of animal, but these cannot always be transferred readily. For example, mice are resistant to infection from prions of hamsters. This means that hamster PrPsc cannot convert mouse PrPc to PrPsc . The situation changes, though, if the mouse PrP gene is replaced by a hamster PrP gene. (This can be done by introducing the hamster PrP gene into the PrP knockout mouse.) A mouse with a hamster PrP gene is sensitive to infection by hamster PrPsc . This suggests that the conversion of cellular PrPc protein into the Sc state requires that the PrPsc and PrPc proteins have matched sequences.
Different “strains” of PrPsc have been distinguished by characteristic incubation periods upon inoculation into mice. This implies that the protein is not restricted solely to alternative states of PrPc and PrPsc but rather that there may be multiple Sc states. These differences must depend on some self-propagating property of the protein other than its sequence. If conformation is the feature that distinguishes PrPc from PrPsc , then there must be multiple conformations, each of which has a self-templating property when it converts PrPsc .
The probability of conversion from PrPc to PrPsc is affected by the sequence of PrP. Gerstmann–Straussler syndrome in humans is caused by a single amino acid change in PrP. This is inherited as a dominant trait. If the same change is made in the mouse PrP gene, mice develop the disease. This suggests that the mutant protein has an increased probability of spontaneous conversion into the Sc state. Similarly, the sequence of the PrP gene determines the susceptibility of sheep to develop the disease spontaneously; the combination of amino acids at three positions (codons 136, 154, and 171) determines susceptibility.
The prion offers an extreme case of epigenetic inheritance, in which the infectious agent is a protein that can adopt multiple conformations, each of which has a self-templating property. This property is likely to involve the state of aggregation of the protein.





















