آخر المواضيع المضافة

 النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية
النبات
الحيوان
الأحياء المجهرية
علم الأمراض
التقانة الإحيائية
التقنية الحيوية المكروبية
التقنية الحياتية النانوية
علم الأجنة
الأحياء الجزيئي
علم وظائف الأعضاء
الغدد
المضادات الحيوية| Fatty acid β-oxidation |


|
|
Read More
Date: 7-1-2022
Date: 24-9-2021
Date: 5-1-2022
|
Fatty acid β-oxidation
The major pathway for catabolism of fatty acids is a mitochondrial pathway called β-oxidation, in which two-carbon fragments are successively removed from the carboxyl end of the fatty acyl CoA, producing acetyl CoA, NADH, and FADH2.
1. Long-chain fatty acid transport into mitochondria: After a LCFA enters a cell, it is converted in the cytosol to its CoA derivative by long-chain fatty acyl CoA synthetase (thiokinase), an enzyme of the outer mitochondrial membrane. Because β-oxidation occurs in the mitochondrial matrix, the fatty acid must be transported across the inner mitochondrial membrane that is impermeable to CoA. Therefore, a specialized carrier transports the long-chain acyl group from the cytosol into the mitochondrial matrix. This carrier is carnitine, and this ratelimiting transport process is called the carnitine shuttle (Fig. 1).

Figure 1: Carnitine shuttle. The net effect is that a long-chain (LC) fatty acyl coenzyme A (CoA) is transported from the outside to the inside of mitochondria. AMP = adenosine monophosphate; PPi = pyrophosphate.
a. Translocation steps: First, the acyl group is transferred from CoA to carnitine by carnitine palmitoyltransferase I (CPT-I), an enzyme of the outer mitochondrial membrane. [Note: CPT-I is also known as CAT-I for carnitine acyltransferase I.] This reaction forms an acylcarnitine and regenerates free CoA. Second, the acylcarnitine is transported into the mitochondrial matrix in exchange for free carnitine by carnitine–acylcarnitine translocase. Carnitine palmitoyltransferase 2 (CPT-II, or CAT-II), an enzyme of the inner mitochondrial membrane, catalyzes the transfer of the acyl group from carnitine to CoA in the mitochondrial matrix, thus regenerating free carnitine.
b. Carnitine shuttle inhibitor: Malonyl CoA inhibits CPT-I, thus preventing the entry of long-chain acyl groups into the mitochondrial matrix. Therefore, when fatty acid synthesis is occurring in the cytosol (as indicated by the presence of malonyl CoA), the newly made palmitate cannot be transferred into mitochondria and degraded. [Note: Muscle tissue, although it does not synthesize fatty acids, contains the mitochondrial isozyme of ACC (ACC2), allowing regulation of β-oxidation. The liver contains both isozymes.] Fatty acid oxidation is also regulated by the acetyl CoA/CoA ratio: As the ratio increases, the CoA-requiring thiolase reaction decreases (Fig. 2).
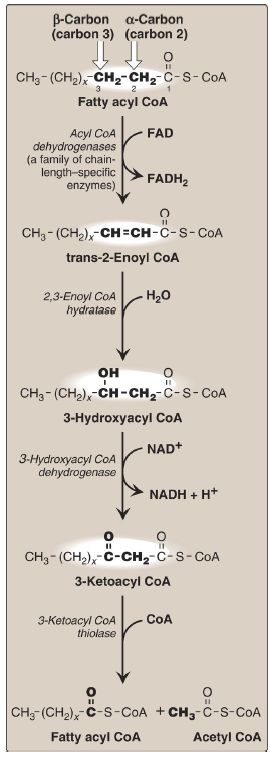
Figure 2: Enzymes involved in the β-oxidation of fatty acyl coenzyme A (CoA). [Note: 2,3-Enoyl CoA hydratase requires a trans double bond between carbon 2 and carbon 3.] FAD(H2) = flavin adenine dinucleotide; NAD(H) = nicotinamide adenine dinucleotide.
c. Carnitine sources: Carnitine can be obtained from the diet, where it is found primarily in meat products. It can also be synthesized from the amino acids lysine and methionine by an enzymatic pathway found in the liver and kidneys but not in skeletal or cardiac muscle. Therefore, these latter tissues are totally dependent on uptake of carnitine provided by endogenous synthesis or the diet and distributed by the blood. [Note: Skeletal muscle contains ~97% of all carnitine in the body.]
d. Carnitine deficiencies: Such deficiencies result in decreased ability of tissues to use LCFA as a fuel. Primary carnitine deficiency is caused by defects in a membrane transporter that prevent uptake of carnitine by cardiac and skeletal muscle and the kidneys, causing carnitine to be excreted. Treatment includes carnitine supplementation. Secondary carnitine deficiency occurs primarily as a result of defects in fatty acid oxidation leading to the accumulation of acylcarnitines that are excreted in the urine, decreasing carnitine availability. Acquired secondary carnitine deficiency can be seen, for example, in patients with liver disease (decreased carnitine synthesis) or those taking the antiseizure drug valproic acid (decreased renal reabsorption). [Note: Defects in mitochondrial oxidation can also be caused by deficiencies in CPT-I and CPT-II. CPT-I deficiency affects the liver, where an inability to use LCFA for fuel greatly impairs that tissue’s ability to synthesize glucose (an endergonic process) during a fast. This can lead to severe hypoglycemia, coma, and death. CPT-II deficiency can affect the liver and cardiac and skeletal muscle. The most common (and least severe) form affects skeletal muscle. It presents as muscle weakness with myoglobinemia following prolonged exercise.
Treatment includes avoidance of fasting and adopting a diet high in carbohydrates and low in fat but supplemented with medium-chain TAG.]
2. Shorter-chain fatty acid entry into mitochondria: Fatty acids ≤12 carbons can cross the inner mitochondrial membrane without the aid of carnitine or the CPT system. Once inside the mitochondria, they are activated to their CoA derivatives by matrix enzymes and are oxidized. [Note: Medium-chain fatty acids are plentiful in human milk. Because their oxidation is not dependent on CPT-I, malonyl CoA is not inhibitory.]
3. β-Oxidation reactions: The first cycle of β-oxidation is shown in Figure 2. It consists of a sequence of four reactions involving the β-carbon (carbon 3) that results in shortening the fatty acid by two carbons at the carboxylate end. The steps include an oxidation that produces FADH2, a hydration, a second oxidation that produces NADH, and a CoAdependent thiolytic cleavage that releases a molecule of acetyl CoA.
Each step is catalyzed by enzymes with chain-length specificity. [Note: For LCFA, the last three steps are catalyzed by a trifunctional protein.] These four steps are repeated for saturated fatty acids of even-numbered carbon chains (n/2) − 1 times (where n is the number of carbons), each cycle producing one acetyl CoA plus one NADH and one FADH2. The final cycle produces two acetyl CoA. The acetyl CoA can be oxidized or used in hepatic ketogenesis (see V. below). The reduced coenzymes are oxidized by the electron transport chain, NADH by Complex I, and FADH2 by coenzyme Q (see p. 75). [Note: Acetyl CoA is a positive allosteric effector of pyruvate carboxylase (see p. 119), thus linking fatty acid oxidation and gluconeogenesis.]
4. β-Oxidation energy yield: The energy yield from fatty acid β-oxidation is high. For example, the oxidation of a molecule of palmitoyl CoA to CO2 and H2O produces 8 acetyl CoA, 7 NADH, and 7 FADH2, from which 131 ATP can be generated. However, activation of the fatty acid requires two ATP. Therefore, the net yield from palmitate is 129 ATP (Fig.3). A comparison of the processes of synthesis and degradation of long-chain saturated fatty acids with an even number of carbon atoms is provided in Figure 4.

Figure 3: Summary of the energy yield from the oxidation of palmitoyl coenzyme A (CoA) (16 carbons). [Note: *Activation of palmitate to palmitoyl CoA requires the equivalent of 2 ATP (ATP → AMP + PPi).] FADH2 = flavin adenine dinucleotide; NADH = nicotinamide adenine dinucleotide; TCA = tricarboxylic acid; CoQ = coenzyme Q; CO2 = carbon dioxide.
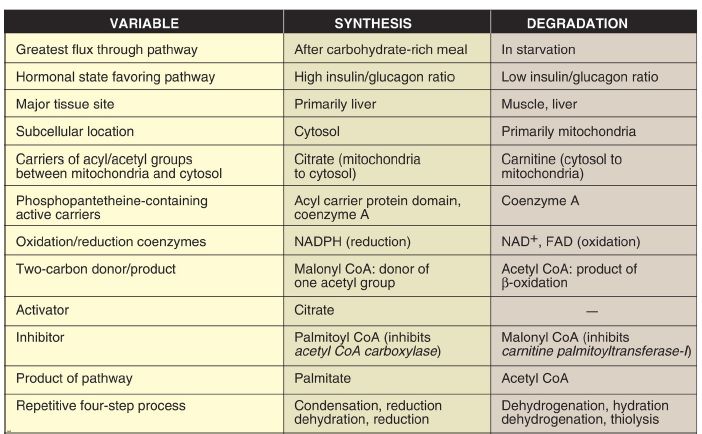
Figure 4: Comparison of the synthesis and degradation of long-chain, evennumbered, saturated fatty acids. NADPH = nicotinamide adenine dinucleotide phosphate; NAD+ = nicotinamide adenine dinucleotide; FAD = flavin adenine dinucleotide; CoA = coenzyme A.
5. Medium-chain fatty acyl CoA dehydrogenase deficiency: Inmitochond ria, there are four fatty acyl CoA dehydrogenase species, each with distinct but overlapping specificity for either short-, medium-, long-, or very-long-chain fatty acids. Medium-chain fatty acyl CoA dehydrogenase (MCAD) deficiency, an autosomal-recessive disorder, is the most common inborn error of β-oxidation, being found in 1:14,000 births worldwide, with a higher incidence in Caucasians of Northern European descent. It results in decreased ability to oxidize fatty acids with six to ten carbons (which accumulate and can be measured in urine), severe hypoglycemia (because the tissues must increase their reliance on glucose), and hypoketonemia (because of decreased production of acetyl CoA). Treatment includes avoidance of fasting.
6. Oxidation of fatty acids with an odd number of carbons: This process proceeds by the same reaction steps as that of fatty acids with an even number of carbons, until the final three carbons are reached. This product, propionyl CoA, is metabolized by a three-step pathway (Fig. 16.20). [Note: Propionyl CoA is also produced during the metabolism of certain amino acids .]
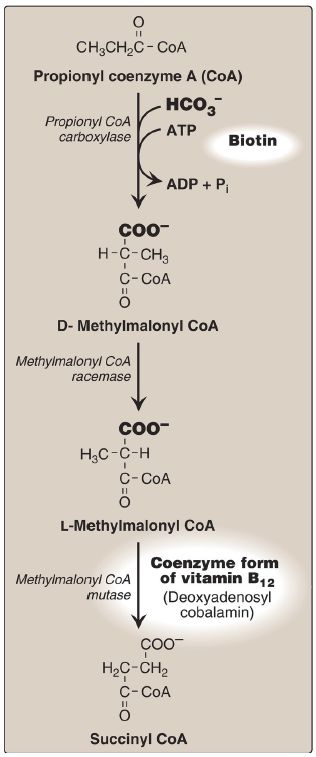
Figure 5: Metabolism of propionyl CoA. ADP = adenosine diphosphate; = bicarbonate; Pi = inorganic phosphate.
a. d-Methylmalonyl CoA synthesis: First, propionyl CoA is carboxylated, forming D-methylmalonyl CoA. The enzyme propionyl CoA carboxylase has an absolute requirement for the coenzymes biotin and ATP, as do ACC and most other carboxylases.
b. l-Methylmalonyl CoA formation: Next, the D-isomer is converted to the L-form by the enzyme methylmalonyl CoA racemase.
c. Succinyl CoA synthesis: Finally, the carbons of L-methylmalonyl CoA are rearranged, forming succinyl CoA, which can enter the TCA cycle . [Note: This is the only example of a glucogenic precursor generated from fatty acid oxidation.] The enzyme methylmalonyl CoA mutase requires a coenzyme form of vitamin B12 (deoxyadenosylcobalamin). The mutase reaction is one of only two reactions in the body that require vitamin B12 . [Note: In patients with vitamin B12 deficiency, both propionate and methylmalonate are excreted in the urine. Two types of heritable methylmalonic acidemia and aciduria have been described: one in which the mutase is missing or deficient (or has reduced affinity for the coenzyme) and one in which the patient is unable to convert vitamin B12 into its coenzyme form. Either type results in metabolic acidosis and neurologic manifestations.]
7. Unsaturated fatty acid β-oxidation: The oxidation of unsaturated fatty acids generates intermediates that cannot serve as substrates for 2,3-enoyl CoA hydratase (see Fig. 2: ). Consequently, additional enzymes are required. Oxidation of a double bond at an odd-numbered carbon, such as 18:1(9) (oleic acid), requires one additional enzyme, 3,2-enoyl CoA isomerase, which converts the 3-cis derivative obtained after three rounds of β-oxidation to the 2-trans derivative required by the hydratase.
Oxidation of a double bond at an even-numbered carbon, such as 18:2(9,12) (linoleic acid), requires an NADPH-dependent 2,4-dienoyl CoA reductase in addition to the isomerase. [Note: Because unsaturated fatty acids are less reduced than saturated fatty acids, fewer reducing equivalents are produced by their oxidation.]
8. Peroxisomal β-oxidation: VLCFA ≥22 carbons in length undergo a preliminary β-oxidation in peroxisomes, because peroxisomes and not mitochondria are the primary site of the synthetase that activates fatty acids of this length. The shortened fatty acid (linked to carnitine) diffuses to a mitochondrion for further oxidation. In contrast to mitochondrial β-oxidation, the initial dehydrogenation in peroxisomes is catalyzed by a FAD-containing acyl CoA oxidase. The FADH2 produced is oxidized by O2, which is reduced to hydrogen peroxide (H2O2). Therefore, no ATP is generated from this step. The H2O2 is reduced to H2O by catalase . [Note: Genetic defects in the ability either to target matrix proteins to peroxisomes (resulting in Zellweger syndrome, a peroxisomal
biogenesis disorder) or to transport VLCFA across the peroxisomal membrane (resulting in X-linked adrenoleukodystrophy) lead to accumulation of VLCFA in the blood and tissues.]

|
|
|
|
التوتر والسرطان.. علماء يحذرون من "صلة خطيرة"
|
|
|
|
|
|
|
مرآة السيارة: مدى دقة عكسها للصورة الصحيحة
|
|
|
|
|
|
|
نحو شراكة وطنية متكاملة.. الأمين العام للعتبة الحسينية يبحث مع وكيل وزارة الخارجية آفاق التعاون المؤسسي
|
|
|