























علم الكيمياء

تاريخ الكيمياء والعلماء المشاهير
التحاضير والتجارب الكيميائية
المخاطر والوقاية في الكيمياء
اخرى
مقالات متنوعة في علم الكيمياء
كيمياء عامة
الكيمياء التحليلية
مواضيع عامة في الكيمياء التحليلية
التحليل النوعي والكمي
التحليل الآلي (الطيفي)
طرق الفصل والتنقية
الكيمياء الحياتية
مواضيع عامة في الكيمياء الحياتية
الكاربوهيدرات
الاحماض الامينية والبروتينات
الانزيمات
الدهون
الاحماض النووية
الفيتامينات والمرافقات الانزيمية
الهرمونات
الكيمياء العضوية
مواضيع عامة في الكيمياء العضوية
الهايدروكاربونات
المركبات الوسطية وميكانيكيات التفاعلات العضوية
التشخيص العضوي
تجارب وتفاعلات في الكيمياء العضوية
الكيمياء الفيزيائية
مواضيع عامة في الكيمياء الفيزيائية
الكيمياء الحرارية
حركية التفاعلات الكيميائية
الكيمياء الكهربائية
الكيمياء اللاعضوية
مواضيع عامة في الكيمياء اللاعضوية
الجدول الدوري وخواص العناصر
نظريات التآصر الكيميائي
كيمياء العناصر الانتقالية ومركباتها المعقدة
مواضيع اخرى في الكيمياء
كيمياء النانو
الكيمياء السريرية
الكيمياء الطبية والدوائية
كيمياء الاغذية والنواتج الطبيعية
الكيمياء الجنائية
الكيمياء الصناعية
البترو كيمياويات
الكيمياء الخضراء
كيمياء البيئة
كيمياء البوليمرات
مواضيع عامة في الكيمياء الصناعية
الكيمياء التناسقية
الكيمياء الاشعاعية والنووية

Anchimeric Assistance
 المؤلف:
William Reusch
المؤلف:
William Reusch
 المصدر:
Virtual Textbook of Organic Chemistry
المصدر:
Virtual Textbook of Organic Chemistry
 الجزء والصفحة:
............
الجزء والصفحة:
............
 10-7-2018
10-7-2018
 3974
3974




+
-
20
Anchimeric Assistance
When the solvolysis rates of alkyl halides and sulfonate esters are measured, some curious influences of neighboring substituents are observed. For example, ethyl chloride, neopentyl chloride (2,2-dimethylpropyl chloride) and 2,2,2-triphenylethyl chloride are all 1º-alkyl chlorides, which hydrolyze in wet formic acid to mixtures of alcohols and olefins (SN1 & E1 mechanisms). The reaction rates for ethyl chloride and neopentyl chloride are nearly identical, but the triphenyl compound reacts 60,000 times faster. Equations for the latter two solvolyses are shown in the following diagram. It is apparent that in both cases an initially formed 1º-carbocation has rearranged prior to product formation, as depicted by clicking on the diagram. However, the increased rate of the phenyl substituted compound is perplexing, especially in view of the greater electronegativity of phenyl groups relative to methyl (note that diphenylacetic acid is over nine times more acidic than isobutyric acid). To explain the unexpected reactivity of 2,2,2-triphenylethyl chloride it is proposed that the pi-electrons of a suitably oriented phenyl group assist the 1º-chloride ionization by bond formation from the side opposite the C-Cl bond, as shown by clicking on the diagram a second time. This intramolecular interaction corresponds to the last example in the previous section, and is similar to an intramolecular SN2 reaction. The resulting phenonium ion would immediately open to a 3º-carbocation, in which the assisting phenyl group has shifted to an adjacent position. In this manner a neighboring aromatic ring accelerates the rate-determining (endothermic) ionization step, an influence called anchimeric assistance (Greek: anchi = neighbor).

The following energy profiles for these reactions illustrate the sequence of events. Both reactions begin by an initial rate-determining ionization step, the transition state of which is colored pink. The activation energy for this step is larger for neopentyl chloride because it leads to a discrete 1º-carbocation. On the other hand, the ionization of triphenylethyl chloride proceeds with assistance from a neighboring phenyl group, and the resulting phenonium ion immediately opens to a very stable diphenyl 3º-carbocation. The second step in the neopentyl chloride solvolysis is a rapid rearrangement of the 1º-carbocation to an isomeric 3º-carbocation. The transition state for this rearrangement is colored green. In both cases, the 3º-carbocation intermediate finally disproportionates to a mixture of substitution and elimination products. The essential difference is that the ionization transition state for neopentyl chloride suffers all the disadvantages associated with the generation of a 1º-carbocation; whereas, the transition state for ionization of triphenylethyl chloride is lowered in energy by its phenonium-like character.

Anchimeric assistance not only manifests itself in enhancement of ionization, but also influences the stereochemical outcome of reactions. The acetolysis of diastereomeric 3-phenyl-2-butanol derivatives provides an example. This alcohol has two chiral centers, and therefore has four stereoisomers in the form of two pairs of enantiomers. The diastereomeric configurations are called erythro and threo, according to their correlation with the tetroses erythrose and threose. As a rule, erythro isomers may assume an eclipsed conformation in which identical or similar substituents on the two stereogenic sites eclipse each other. Threo isomers cannot assume such a conformation. In the following diagram, a tosylate derivative of one enantiomer of each diastereomer is drawn as a Fischer projection. These isomers were solvolyzed in hot acetic acid solution, buffered with sodium acetate, and the configurations of the resulting acetate esters were determined. As expected from a SN1 process, some E1 elimination product was also obtained. Remarkably, each diastereomer is converted to its equivalent diastereomeric acetate (retention of configuration). Furthermore,the erythro compound retains its enantiomeric purity; whereas the threo tosylate gives racemic acetate and is itself racemized during reaction. If an open carbocation intermediate were formed in these reactions, mixtures of erythro and threo acetates would be expected from both tosylates, but only trace amounts of the opposite diastereomer were found among the products.
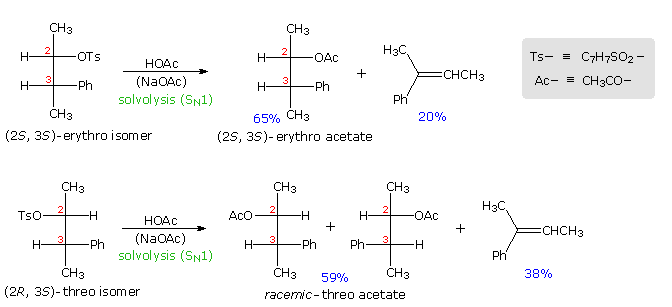
By clicking on the diagram the controlling influence of phenyl group anchimeric assistance will be demonstrated. First, the molecule assumes a conformation in which the phenyl substituent is oriented anti to the tosylate group. Next, a pair of pi-electrons from the benzene ring bonds to C2 as the tosylate anion departs, generating a phenonium intermediate (in brackets). The intermediate from the erythro tosylate is chiral, but that from the threo tosylate is achiral (note the plane of symmetry bisecting the three-membered ring). In each case C2 & C3 are constitutionally equivalent, and nucleophilic attack by acetate anion takes place equally well at either position (green and light blue arrows). As a result of equal rates of product formation by acetate bonding to C2 & C3, the achiral threo intermediate yields a 50:50 (racemic) mixture of threo enantiomers: (2R,3S) from the blue arrows and (2S, 3R) from the green arrows. In contrast, acetate bonding to C2 & C3 of the erythro intermediate produces the same enantiomer of the erythro product (2S,3S). Since the initial ionization to phenonium intermediates is reversible, we are not surprised to find that unreacted erythro tosylate is unchanged; whereas, unreacted threo tosylate is racemized.
 الاكثر قراءة في مواضيع عامة في الكيمياء العضوية
الاكثر قراءة في مواضيع عامة في الكيمياء العضوية
 اخر الاخبار
اخر الاخبار
اخبار العتبة العباسية المقدسة

الآخبار الصحية


 قسم الشؤون الفكرية يصدر كتاباً يوثق تاريخ السدانة في العتبة العباسية المقدسة
قسم الشؤون الفكرية يصدر كتاباً يوثق تاريخ السدانة في العتبة العباسية المقدسة "المهمة".. إصدار قصصي يوثّق القصص الفائزة في مسابقة فتوى الدفاع المقدسة للقصة القصيرة
"المهمة".. إصدار قصصي يوثّق القصص الفائزة في مسابقة فتوى الدفاع المقدسة للقصة القصيرة (نوافذ).. إصدار أدبي يوثق القصص الفائزة في مسابقة الإمام العسكري (عليه السلام)
(نوافذ).. إصدار أدبي يوثق القصص الفائزة في مسابقة الإمام العسكري (عليه السلام)



















